登陆有奖并可浏览互动!
您需要 登录 才可以下载或查看,没有账号?立即注册


×
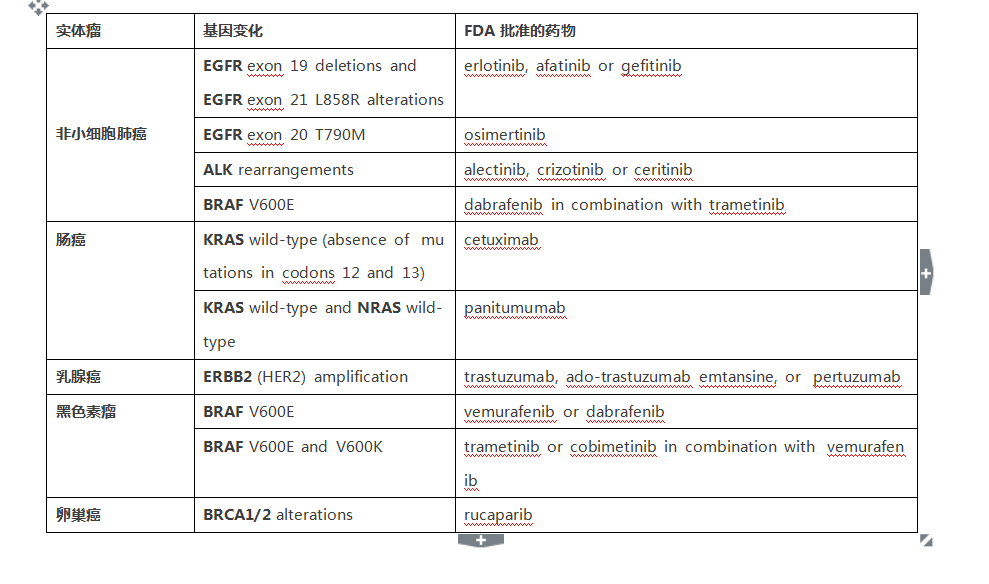
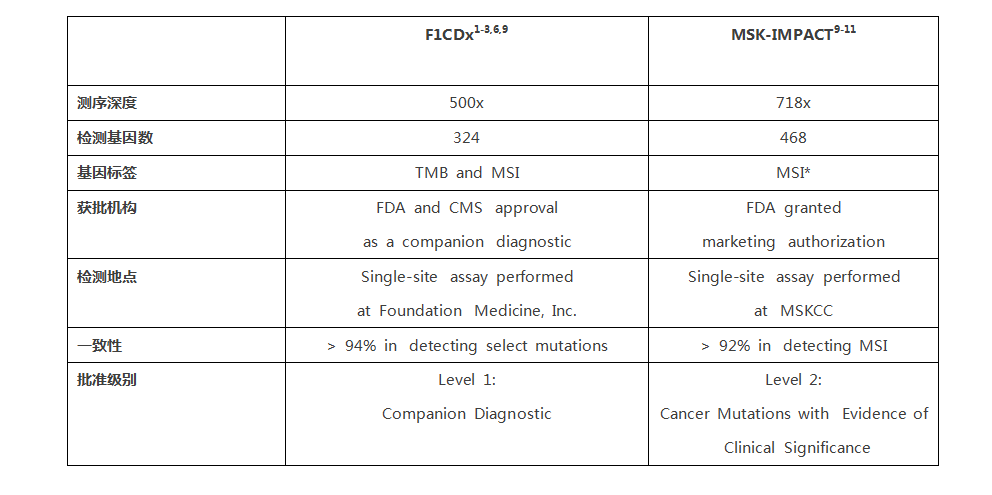
2017年11月30日肿瘤届的一个大事件,NGS被FDA批准为肿瘤检测伴随诊断!),Foundation Medicine的F1CDx作为实体瘤的伴随诊断FDA被获批。他能够检测324个肿瘤相关基因,以及2个肿瘤相关标签(TMB和MSI),这改变了目前一种检测针对一种肿瘤治疗药物的格局,是一个里程碑式的事件。 首先我们来看看F1CDx的应用 F1CDx作为了FDA批准的系列药物在五种实体瘤的伴随诊断 2. 这种测序可以影响治疗决策帮助选择包含免疫治疗在内的肿瘤用药 3. FDA的获批支持及大大促进了了肿瘤精准医疗的发展,改变了“一瘤一诊一药”的格局。 另外,我们知道MSI的状态也是很重要的指导免疫治疗的标记物,虽然该平台未被作为肿瘤MSI状态的伴随诊断,但也能提供了依据指导肿瘤免疫治疗药物的选择。 今年11月中旬,我们看到了FDA批准了MSKCC的的二代测序平台被FDA批准作为肿瘤检测的补充诊断。该平台可以检测468个基因突变,及其他分子变化。这可以说是一次重要事件,但为啥说F1CDx是里程碑? 因为,F1CDx是伴随诊断,包括插入缺失、点突变、重排在内的基因变化检测,还有患者的TMB及MSI状况。FDA批准及美国CMS医保覆盖,针对五种实体瘤的具体药物使用,直接关联到患者用药及临床结局。 而MSKCC-IMPACT平台同样做为体外诊断被FDA批准和非伴随诊断,只是510(k)批准,要比伴随诊断简单得多,并没有和任何药物的使用相关联。 二者对比: 美国FDA按照IVD(体外诊断产品)的危险性和复杂程度将其分为三类: I类风险较低,有医疗保险和医疗不住服务中心一句CLIA对临床实验室和科研实验室监管; II类风险中,按照FDA上市前同时510(k)流程对申报产品及同类产品进行“等效性”对比。 III类风险程度对高,需通过上市前审批流程(PMA)批准,比起II类严格得多,需要通过一定规模的临床试验证实其安全性和有效性。例如伴随诊断。
来源:搜狐新闻
|

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 






 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 发表于 2017-12-21 22:53
发表于 2017-12-21 22:53