金桔
金币
威望
贡献
回帖0
精华
在线时间 小时
|
登陆有奖并可浏览互动!
您需要 登录 才可以下载或查看,没有账号?立即注册


×
对核酸分子(如DNA或RNA)进行测序,可以得到有关其核苷酸(腺嘌呤、胸腺嘧啶、胞嘧啶和鸟嘌呤)顺序的信息,这些核苷酸提供了生命所需的遗传指令。
测序技术的发展源于Walter Fiers、Frederick Sanger和Ray Wu在1970年代的开创性工作[1]。在几十年间,最初用于核酸测序的方法和技术经历了一个迅猛的发展:从读取单个RNA分子开始,对整个生物体的基因组进行测序成为可能[2]。
第一个人类基因组草案于2001年在人类基因组计划内公布[3],并与两年后完成。
商业DNA测序仪首次生产于20世纪90年代末,它们被命名为第二代或第二代测序仪,以区别于第一批实验性测序仪,并允许对整个基因组进行测序。
在接下来的20年里,测序仪经历了快速演变,在不同程度上优化了速度、准确性、测序深度和读长等参数[4]。
各种各样的测序方法已被开发出来用于特定的应用。虽然相对较新,但最有趣的方法之一是单细胞测序。本文探讨了这项技术是如何工作的,以及它能够给我们提供什么信息。
1、什么是单细胞测序,什么是单细胞RNA测序?
传统的第二代测序(NGS)检查一个细胞群的基因组,如细胞培养、组织、器官或整个生物体。它的输出是细胞群的「平均基因组」,而单细胞测序测量的是细胞群中单个细胞的基因组[5]。现在,传统方法因此被称为批量测序,以区别于单细胞技术。
目前,单细胞测序技术可用于测量一个群体中每个细胞的基因组(scDNA-seq)、DNA-甲基组或转录组(scRNA-seq)。
这些技术已被用于识别癌细胞中的新突变,探索胚胎发育过程中发生的渐进式表观基因组变化,并评估一个看似同质的细胞群是如何表达特定基因的(图1)[6]。
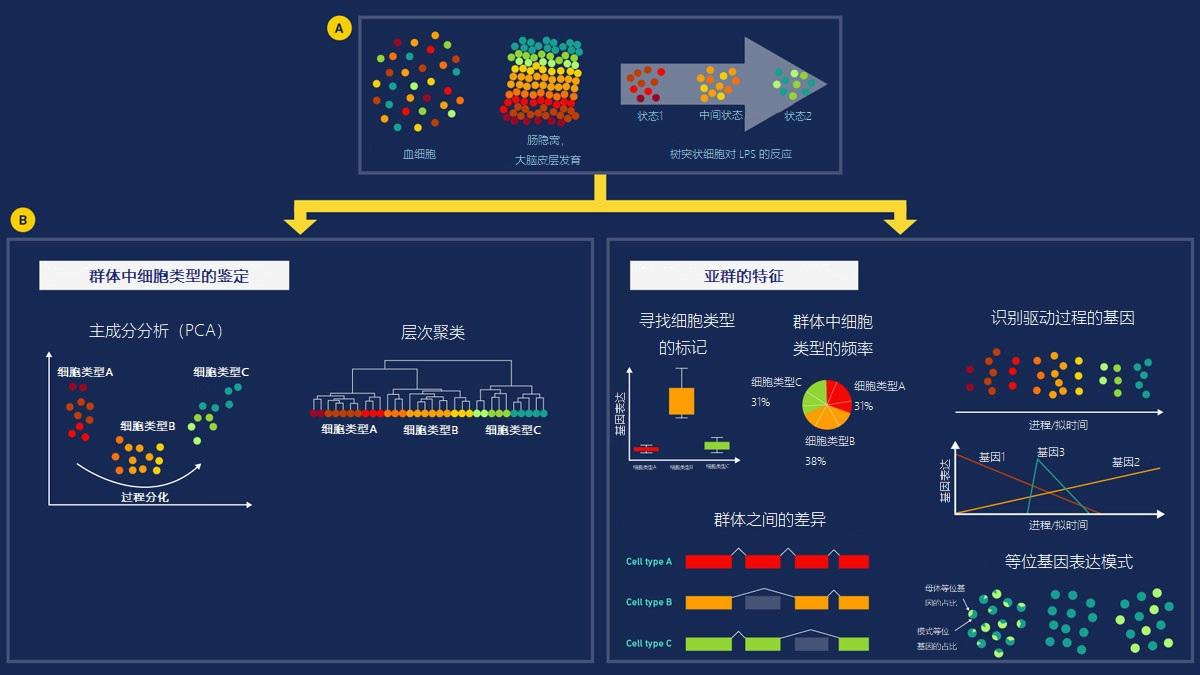
图1 | 利用单细胞测序技术,我们可以在一个看似同质的细胞群中确定新的亚群或细胞状态。A)显示了一个细胞群可能表现出异质性的不同方式。B)显示了细胞群中的细胞类型是如何被识别和表征的。
2、单细胞测序是如何工作的?
所有单细胞测序技术需要四个主要步骤(图2)。
- 从细胞群中分离出单细胞。
- 提取、处理和扩增每个分离的细胞的遗传物质。
- 制备包括分离细胞遗传物质的「测序库」。
- 使用第二代测序仪对该库进行测序。
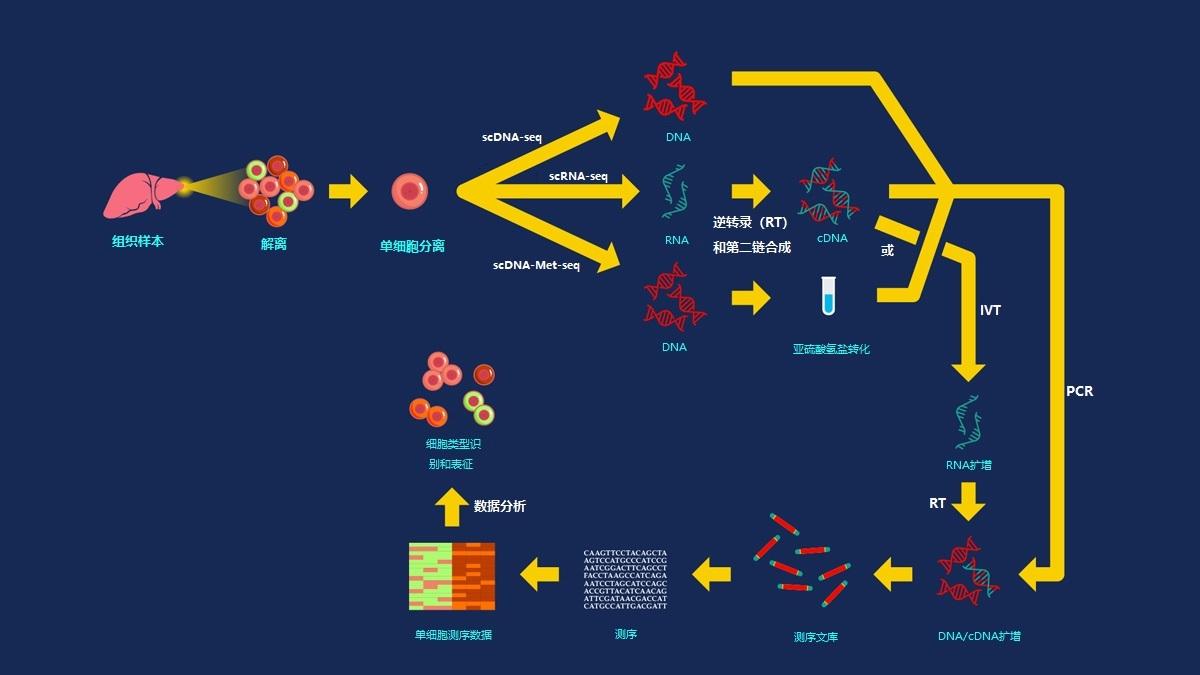
图2 | 单细胞测序工作流程,根据所关注的遗传物质,样品制备过程略有不同。然而,在所有情况下,产生的测序材料是每个细胞的条形码DNA或cDNA库,然后可以在选择的测序平台上测序。
2.1、细胞的分离和基础样品处理
细胞可以用不同的方法进行分离[7,8],其选择主要取决于样品的性质和细胞分离后需要的处理步骤。
每种方法的性能由其效率(每单位时间可分离多少细胞)、纯度(收集到的目标细胞的比例)和回收率(收集到的目标细胞与最初可用的目标细胞总数相比的比例)决定。下面,让我们介绍一些最常用的技术[9]:
- 荧光激活细胞分选(FACS):包括用附着在目标特异性抗体上的荧光分子来标记细胞选择所依据的细胞蛋白。细胞内的标记物可以通过细胞通透性来获取。因此,该技术允许根据多个参数来选择细胞。然而,FACS需要>10,000个起始细胞,而且快速流动可能会损害细胞的生存能力[7];
- 磁激活细胞分选(MACS):使用抗体介导的超顺磁纳米粒子来标记目标细胞上的特定蛋白质。然而,这意味着,与FACS不同,只有细胞表面分子可以作为标记活细胞的目标[10],然后使用外部磁场来隔离被标记的细胞,而其他细胞则被洗掉。因此,MACS分离的纯度取决于用于标记的抗体的特异性和亲和力;
- 激光捕获显微切割(LCM):使用激光从放置在显微镜玻片上的固体组织样本中分离出目标细胞。隔离可以通过两种方式进行:直接,当红外光将激光能量转移到只与目标细胞特异性结合的热可塑性聚合物上时;间接,当紫外光消融细胞时。与FACS和MACS不同,LCM可用于完整的组织。它也是快速和可靠的。然而,LCM需要通过目视检查其形态来识别目标细胞。另外,细胞在分离过程中可能被切片,紫外线可能会损害DNA和RNA分子[11];
- 手工挑选细胞:又被称为微操作,需要一个倒置的显微镜,结合微吸管来选择和分离目标细胞。微操作已经被用于活体培养和胚胎细胞。然而,它的产量是有限的,而且与LCM一样,该技术需要熟练的专业人员来正确识别目标细胞[12]。
在进行文库制备之前,要对细胞分离的质量进行评估,并通过成像评估细胞的活力。
RNA的完整性也可以被评估,这对scRNA-seq分析特别重要。然后对分离的细胞进行裂解,相关的遗传物质(DNA或RNA)被分离和扩增,以提供足够的后续检测,因为单细胞通常只产生极少量的DNA或RNA。
这些步骤产生的材料是单链DNA,甚至用于scRNA-seq分析,我们将在下文中进行解释。已经制定了一些方案来处理特定研究的要求和限制,例如在可用细胞数量少的情况下[13]。
2.2、测序文库的制备
为了对扩增的DNA进行测序,首先需要将其制成一个测序文库[13,14]。测序文库是来自一个细胞群的单链DNA片段的集合,或者在单细胞测序的情况下,来自一个特定的细胞。
扩增后,DNA片段被唯一的条码化,以识别它们属于哪个起始细胞,并在5'和3'端添加特定的适配序列。
需要测序的DNA部分通常被命名为插入物,条形码和适配体在插入物的一端或两端加盖,属于同一测序库的所有DNA片段都使用相同的寡核苷酸序列进行条码化。
这允许在同一测序过程中把不同的文库集中在一起进行测序,适配体取决于平台,需要对片段进行测序。所有的样品和文库制备步骤都有商业试剂盒。为了确保得到的文库准确地再现原始细胞的状态,有几种质量控制的方式(图3)[15]。
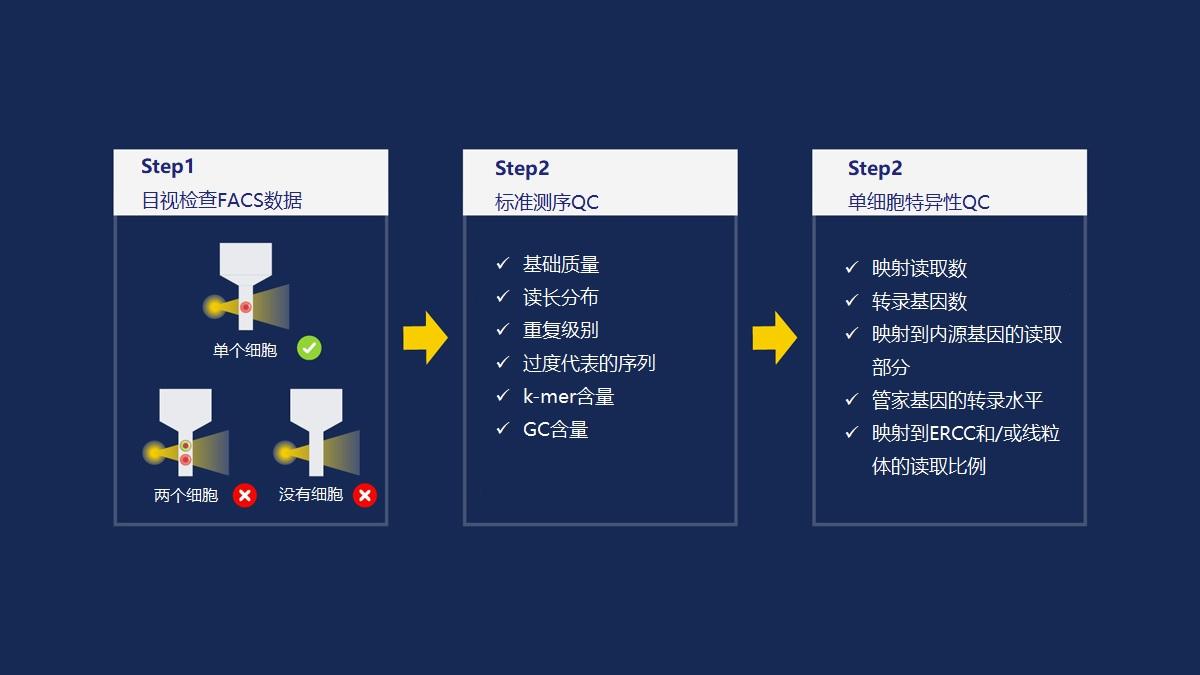
图3 | 评估文库制备和测序结果的质量控制,在这个例子中,单细胞是通过FACS方法分离出来的。
2.3、测序
各种商业上可用的测序平台已经开发出略有不同的方法。在此,我们重点讨论通过合成方法进行测序,包括火法测序(pyrosequencing)和可逆终止器测序等变化。
在测序之前,一个扩增步骤通常会产生一组DNA片段克隆(通常通过桥式扩增或乳化PCR)。由于每组克隆在测序过程中发出相同的信号,因此产生的簇或孔信号足够强,可以进行检测[16]。
这种类型的测序通常在一个芯片内进行,它可能包含微孔。适配体和其他分子,如聚合酶,被绑定在芯片上(或微孔的底部),并与连接在插入物上的适配体相互作用。
然后,插入物测序需要多个复制步骤,由聚合酶执行,并使用荧光标记的核苷酸。
在每个周期中,加入一个荧光标记的核苷酸,如果被聚合酶结合,就会触发特定核苷酸的光发射。
在下一个周期开始前,所有片段同时发出的光谱通过摄像机被记录下来。当每个核苷酸发出不同的光时,测序仪就会逐个周期地重建所有插入物的序列。测序仪还读取插入物的标签,将每个测量结果分配到相应的库中。
质子检测测序(Proton detection sequencing)使用一种不同的合成测序方法。片段通常被绑定在珠子上,并被扩增以覆盖每个珠子(类似于热测序)。然而,当碱基在测序过程中被添加时,它不是一个荧光标签和光释放,而是释放一个质子,然后可以被检测和记录。
另一种不太常见的测序方法是结扎法测序(sequencing by ligation)。这种方法使用DNA连接酶而不是DNA聚合酶,连接荧光标记的短序列而不是核苷酸。
在测序之前,通常使用乳剂PCR扩增化学方法对DNA片段进行扩增。由于插入物的每个核苷酸随后被测序两次,这种方法提供非常准确的读数。然而,结扎测序只能输出较短的读数,而且与回文序列不兼容。
3、单细胞测序的类型
单细胞测序技术可以测量不同类型的遗传物质,单细胞的基因组、转录组或甲基组。本部分内容解释了样品制备的主要差异(图2)和这三种测序亚型的最相关应用。
3.1、单细胞基因组测序
通过确定单细胞的基因组,scDNA-seq可以对细胞群的基因组异质性进行研究[17]。
因此,它主要用于研究微生物组和癌症。微生物组是单细胞生物的社区,scDNA-seq测量其微生物成分的基因组,而不需要先分离和培养它们。
然后,测序数据可用于研究微生物组的组成,从而研究其生态学、进化和改变[18,19]。
在癌症研究中,scDNA-seq被用来研究肿瘤内的遗传异质性或识别新型致癌突变[20,21]。由于这些创新能力,scDNA-seq大大促进了精准医疗的发展[22]。
对于scDNA-seq,从分离的细胞中提取的DNA最常使用多重置换扩增法(MDA)或多重退火和循环扩增循环法(MALBAC)[23]。
MDA允许快速扩增微量的DNA,提供一个优秀但不均匀的基因组覆盖。另一方面,MALBAC提供的基因组覆盖率较低但更均匀,因此更适合检测拷贝数变异。
如前所述,库是由扩增的DNA生成的。为了使这种方法成功,均匀和有效的扩增是至关重要的。
然而,没有一种扩增方法是无懈可击的。特别是检测单核苷酸多态性(SNPs)和拷贝数变异的效率往往很低。因此,不同的扩增方法已经被开发出来,以改善对特定突变类型的检测[24]。
3.2、单细胞转录组测序(单细胞RNA测序,单细胞RNA-seq,或scRNA seq)
scRNA-seq测量给定样本中每个细胞内的RNA分子。这一信息提供了细胞收获时转录组(正在转录的基因)的快照[25,26]。
自从开发以来,scRNA-seq已经发现了大量的应用,虽然一个基因表达的最终产品是蛋白质,但检测其信使RNA(mRNA)表明该基因被打开,因此,有可能随后被翻译和表达。
此外,共同转录的基因可用于推断特定细胞表型所依据的基因调控网络[27],细胞群的转录差异可以帮助识别亚群,如肿瘤块的恶性细胞[28]。
scRNA-seq也被用来研究重要的基因转录特征,如剪接模式和单倍体基因转录[29,30]。由于细菌细胞中mRNA拷贝数低和RNA不稳定等因素,原核生物scRNA-seq一直是个问题。然而,这些问题正在逐渐被克服[31,32]。
分离的细胞被裂解,它们的mRNA分子是多腺苷酸化的,用poly[T]-primers来富集。
这是一个关键步骤,因为细胞中的大多数RNA分子是核糖体(rRNA):非常大,而且通常不是转录组测序的目标。
接下来,逆转录酶将聚[T]引物、单链mRNA转换成互补DNA(cDNA)。然后用PCR或体外转录(IVT)来扩增cDNA分子。
最后,条形码标签和测序平台要求的其他短序列被添加到cDNA分子中[26]。
这种技术的测序质量受一些因素的影响,主要是可以从一个细胞群中得到的文库总数和检测到的读数。
理想的细胞数量取决于不同细胞亚群或状态的预期数量。读数的数量表明一个转录组被测序的深度,这取决于基因组的大小,更高的读数深度提供更可靠的细节,样品和文库制备方案也会影响结果的质量。
3.3、单细胞DNA甲基组测序
DNA甲基化涉及到将一个甲基转移到一个胞嘧啶碳(通常是C5)上。甲基化是一种表观遗传机制,它改变了DNA的活性而不影响其序列,当在基因启动子中时,DNA甲基化通常会抑制该基因的转录[33]。
因此,单细胞DNA甲基组测序(scDNA-Met-seq)可以用来研究在一个原本基因相同的细胞群中的表观遗传变化,引起不同的表型。
DNA甲基化对细胞身份也至关重要,是X染色体失活、基因组印记、可转录元素的压制、衰老和致癌的关键。这项技术主要用于发育研究[34],但也被用于探索罕见的和极其活跃的肿瘤细胞亚群[35]。
scDNA-Met-seq的样本和文库制备与scDNA-seq类似,但多了一个步骤。在扩增之前,DNA要经过亚硫酸氢盐处理,它只将非甲基化的胞嘧啶残基转化为尿嘧啶,而5-甲基胞嘧啶残基则不受影响[36]。
然而,亚硫酸氢盐处理往往会使DNA碎片化和降解。其他方法,如甲基化敏感的限制性酶,已经被探索过,但仍不能用于单细胞测序。
4、分析单细胞测序数据
测序仪的最终原始输出首先在测序机内直接处理,返回二进制碱基调用(BCL)文件和质量分数。
BCL文件是二进制格式的原始测序输出,为了进一步分析,BCL文件然后被转换成FASTQ文件,一个包含序列信息和质量分数的文本文件(图3)。
这一步骤通常是在使用条形码标签对来自不同库的数据进行解复用后,在Linux服务器上进行。
可以对FASTQ文件进行处理,使序列与模板基因组相一致,对其进行注释,检测变体,进行差异转录分析,并将数据可视化[37,38]。
一些第三方学术机构开发的脚本可用于执行这些初步分析[39,40],鉴于FASTQ数据文件的大小(通常每个10~200G),这些分析的计算成本很高,因此,通常在Linux服务器上使用其命令行进行。
由此产生的数据可以使用数据分析和统计工具进一步调查,如数据归一化、主成分分析、t分布的随机邻居嵌入分析、聚类分析和路径或基因组富集分析。
这些工具在探索大型数据集时很有帮助,因为它们可以识别出意想不到的模式和生物行为,以及最显著地驱动特定表型的基因或转录物。
特别是,Bioconductor是一个为R统计编程语言开发的杂项包,为基因组学数据的分析提供免费的开源软件[41]。该软件包内的工具被设计用来进行上述分析并将其结果可视化。某些工作流程和功能已经专门为单细胞测序分析进行了优化[42,43]。
5、单细胞测序的作用
一个组织或器官内的每个细胞以不同的方式对整个机体的生理/病理做出贡献。
利用单细胞技术,我们可以探测每个细胞,并测量其对整个细胞群,以及其有机体或生态系统的具体贡献。
这种独特的细节水平在研究稀有细胞或探索同细胞类型群体中的表型变化时特别有价值。
例如,scRNA-seq已被用于研究罕见的抗原特异性T或B细胞[44],测量人类微生物组的组成和结构[45],研究抗化疗肿瘤亚群的起源和发展[46],发现植物组织中以前未知的基因功能[47],研究肿瘤进展机制并根据肿瘤内细胞异质性进行预后预测[48]。
由于单细胞测序技术的独特性,这些以及更多的应用已经在各个领域成为可能。
6、单细胞测序技术和其他技术的联合使用
现在,各种全息技术经常被结合起来,研究单细胞的多层次状态[49,50]。通过结合之前描述的测序技术,有可能研究同一细胞群中的基因组、表观基因组和转录组景观[51,52]。
测序技术也经常与蛋白质组学方法相结合,包括代谢组学、磷蛋白组学、乙酰组学和糖蛋白组学[53,54]。结合不同的单细胞全貌学方法,可以更深入地了解细胞群的异质性,也可以识别更多的亚群,因为其他技术可以发现不同类型的变化。
也有可能推断出一种全息技术观察到的变化与另一种技术观察到的变化之间的功能联系。这些信息可以帮助确定新的因果关系,从而确定一个已知表型背后的机制。
已经有一些计算方法被开发出来来整合不同的全能基因数据集[55,56],包括创新的、基于机器学习的方法[57]。
然而,整合多组学单细胞数据的算法往往仍不充分。虽然文库制备方案和测序技术已经非常完善,但数据分析工具已经滞后,现在可能构成了这个领域最大的挑战。
诊断科学编辑团队收集、整理和编撰,如需更多咨询,请关注公众号 诊断科学(DiagnosticsScience)。
参考文献
- Jou WM, Haegeman G, Ysebaert M, Fiers W. Nucleotide sequence of the gene coding for the bacteriophage MS2 coat protein. Nature. 1972;237(5350):82-88. doi:10.1038/237082a0
- Sanger F, Air GM, Barrell BG, et al. Nucleotide sequence of bacteriophage φX174 DNA. Nature. 1977;265(5596):687-695. doi:10.1038/265687a0
- Venter JC, Adams MD, Myers EW, et al. The sequence of the human genome. Science. 2001;291(5507):1304-1351. doi:10.1126/science.1058040
- Liu L, Li Y, Li S, et al. Comparison of next-generation sequencing systems. J Biomed Biotechnol. 2012;2012:1-11. doi:10.1155/2012/251364
- Tang X, Huang Y, Lei J, Luo H, Zhu X. The single-cell sequencing: new developments and medical applications. Cell Biosci. 2019;9(1):53. doi:10.1186/s13578-019-0314-y
- Mannarapu M, Dariya B, Bandapalli OR. Application of single-cell sequencing technologies in pancreatic cancer. Mol Cell Biochem. 2021;476(6):2429-2437. doi:10.1007/s11010-021-04095-4
- Gross A, Schoendube J, Zimmermann S, Steeb M, Zengerle R, Koltay P. Technologies for single-cell isolation. Int J Mol Sci. 2015;16(8):16897-16919. doi:10.3390/ijms160816897
- Valihrach L, Androvic P, Kubista M. Platforms for single-cell collection and analysis. Int J Mol Sci. 2018;19(3):807. doi:10.3390/ijms19030807
- Hu P, Zhang W, Xin H, Deng G. Single cell isolation and analysis. Front Cell Dev Biol. 2016;4. doi:10.3389/fcell.2016.00116
- Welzel G, Seitz D, Schuster S. Magnetic-activated cell sorting (MACS) can be used as a large-scale method for establishing zebrafish neuronal cell cultures. Sci Rep. 2015;5(1):7959. doi:10.1038/srep07959
- Espina V, Wulfkuhle JD, Calvert VS, et al. Laser-capture microdissection. Nat Protoc. 2006;1(2):586-603. doi:10.1038/nprot.2006.85
- Citri A, Pang ZP, Südhof TC, Wernig M, Malenka RC. Comprehensive qPCR profiling of gene expression in single neuronal cells. Nat Protoc. 2012;7(1):118-127. doi:10.1038/nprot.2011.430
- Hess JF, Kohl TA, Kotrová M, et al. Library preparation for next generation sequencing: A review of automation strategies. Biotechnol Adv. 2020;41:107537. doi:10.1016/j.biotechadv.2020.107537
- Syed F, Grunenwald H, Caruccio N. Next-generation sequencing library preparation: simultaneous fragmentation and tagging using in vitro transposition. Nat Methods. 2009;6(11):i-ii. doi:10.1038/nmeth.f.272
- Endrullat C, Glökler J, Franke P, Frohme M. Standardization and quality management in next-generation sequencing. Appl Transl Genom. 2016;10:2-9. doi:10.1016/j.atg.2016.06.001
- Pareek CS, Smoczynski R, Tretyn A. Sequencing technologies and genome sequencing. J Appl Genet. 2011;52(4):413-435. doi:10.1007/s13353-011-0057-x
- Gawad C, Koh W, Quake SR. Single-cell genome sequencing: current state of the science. Nat Rev Genet. 2016;17(3):175-188. doi:10.1038/nrg.2015.16
- Kashtan N, Roggensack SE, Rodrigue S, et al. Single-cell genomics reveals hundreds of coexisting subpopulations in wild prochlorococcus. Science. 2014;344(6182):416-420. doi:10.1126/science.1248575
- Swan BK, Tupper B, Sczyrba A, et al. Prevalent genome streamlining and latitudinal divergence of planktonic bacteria in the surface ocean. PNAS. 2013;110(28):11463-11468. doi:10.1073/pnas.1304246110
- Zhang L, Dong X, Lee M, Maslov AY, Wang T, Vijg J. Single-cell whole-genome sequencing reveals the functional landscape of somatic mutations in B lymphocytes across the human lifespan. PNAS. 2019;116(18):9014-9019. doi:10.1073/pnas.1902510116
- Francis JM, Zhang CZ, Maire CL, et al. EGFR variant heterogeneity in glioblastoma resolved through single-nucleus sequencing. Cancer Discov. 2014;4(8):956-971. doi:10.1158/2159-8290.CD-13-0879
- Wiedmeier JE, Noel P, Lin W, von Hoff DD, Han H. Single-cell sequencing in precision medicine. Cancer Treat Res. Published online 2019:237-252. doi:10.1007/978-3-030-16391-4_9
- Chen M, Song P, Zou D, et al. Comparison of multiple displacement amplification (MDA) and multiple annealing and looping-based amplification cycles (MALBAC) in single-cell sequencing. PLoS ONE. 2014;9(12):e114520. doi:10.1371/journal.pone.0114520
- Treff NR, Su J, Tao X, Northrop LE, Scott RT. Single-cell whole-genome amplification technique impacts the accuracy of SNP microarray-based genotyping and copy number analyses. Basic Sci Repro Med. 2011;17(6):335-343. doi:10.1093/molehr/gaq103
- Kolodziejczyk AA, Kim JK, Svensson V, Marioni JC, Teichmann SA. The technology and biology of single-cell RNA sequencing. Mol Cell. 2015;58(4):610-620. doi:10.1016/j.molcel.2015.04.005
- Haque A, Engel J, Teichmann SA, Lönnberg T. A practical guide to single-cell RNA-sequencing for biomedical research and clinical applications. Genome Med. 2017;9(1):75. doi:10.1186/s13073-017-0467-4
- Wagner A, Regev A, Yosef N. Revealing the vectors of cellular identity with single-cell genomics. Nat Biotechnol. 2016;34(11):1145-1160. doi:10.1038/nbt.3711
- Tirosh I, Izar B, Prakadan SM, et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016;352(6282):189-196. doi:10.1126/science.aad0501
- Shalek AK, Satija R, Adiconis X, et al. Single-cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature. 2013;498(7453):236-240. doi:10.1038/nature12172
- Deng Q, Ramsköld D, Reinius B, Sandberg R. Single-cell RNA-seq reveals dynamic, random monoallelic gene expression in mammalian cells. Science. 2014;343(6167):193-196. doi:10.1126/science.1245316
- Imdahl F, Vafadarnejad E, Homberger C, Saliba A-E, Vogel J. Single-cell RNA-sequencing reports growth-condition-specific global transcriptomes of individual bacteria. Nat Microbiol. 2020;5(10):1202-1206. doi:10.1038/s41564-020-0774-1
- Kuchina A, Brettner LM, Paleologu L, et al. Microbial single-cell RNA sequencing by split-pool barcoding. Science. 2021;371(6531):eaba5257. doi:10.1126/science.aba5257
- Moore LD, Le T, Fan G. DNA methylation and its basic function. Neuropsychopharmacology. 2013;38(1):23-38. doi:10.1038/npp.2012.112
- Zhu P, Guo H, Ren Y, et al. Single-cell DNA methylome sequencing of human preimplantation embryos. Nat Genet. 2018;50(1):12-19. doi:10.1038/s41588-017-0007-6
- Gkountela S, Castro-Giner F, Szczerba BM, et al. Circulating tumor cell clustering shapes DNA methylation to enable metastasis seeding. Cell. 2019;176(1-2):98-112.e14. doi:10.1016/j.cell.2018.11.046
- Smallwood SA, Lee HJ, Angermueller C, et al. Single-cell genome-wide bisulfite sequencing for assessing epigenetic heterogeneity. Nat Methods. 2014;11(8):817-820. doi:10.1038/nmeth.3035
- Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15-21. doi:10.1093/bioinformatics/bts635
- Trapnell C, Roberts A, Goff L, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7(3):562-578. doi:10.1038/nprot.2012.016
- Bray NL, Pimentel H, Melsted P, Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol. 2016;34(5):525-527. doi:10.1038/nbt.3519
- Bankevich A, Nurk S, Antipov D, et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19(5):455-477. doi:10.1089/cmb.2012.0021
- Lawrence M, Huber W, Pagès H, et al. Software for computing and annotating genomic ranges. PLoS Comput Biol. 2013;9(8):e1003118. doi:10.1371/journal.pcbi.1003118
- Amezquita RA, Lun ATL, Becht E, et al. Orchestrating single-cell analysis with Bioconductor. Nat Methods. 2020;17(2):137-145. doi:10.1038/s41592-019-0654-x
- Perraudeau F, Risso D, Street K, Purdom E, Dudoit S. Bioconductor workflow for single-cell RNA sequencing: Normalization, dimensionality reduction, clustering, and lineage inference. F1000Research. 2017;6:1158. doi:10.12688/f1000research.12122.1
- Shalek AK, Satija R, Adiconis X, et al. Single-cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature. 2013;498(7453):236-240. doi:10.1038/nature12172
- Balachandran M, Cross KL, Podar M. Single-cell genomics and the oral microbiome. J Dent Res. 2020;99(6):613-620. doi:10.1177/0022034520907380
- Kim C, Gao R, Sei E, et al. Chemoresistance evolution in triple-negative breast cancer delineated by single-cell sequencing. Cell. 2018;173(4):879-893.e13. doi:10.1016/j.cell.2018.03.041
- Wendrich JR, Yang B, Vandamme N, et al. Vascular transcription factors guide plant epidermal responses to limiting phosphate conditions. Science. 2020;370(6518). doi:10.1126/science.aay4970
- Patel AP, Tirosh I, Trombetta JJ, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344(6190):1396-1401. doi:10.1126/science.1254257
- Lake BB, Chen S, Sos BC, et al. Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain. Nat Biotechnol. 2018;36(1):70-80. doi:10.1038/nbt.4038
- Granja JM, Klemm S, McGinnis LM, et al. Single-cell multiomic analysis identifies regulatory programs in mixed-phenotype acute leukemia. Nat Biotechnol. 2019;37(12):1458-1465. doi:10.1038/s41587-019-0332-7
- Kashima Y, Sakamoto Y, Kaneko K, Seki M, Suzuki Y, Suzuki A. Single-cell sequencing techniques from individual to multiomics analyses. Exp Mol Med. 2020;52(9):1419-1427. doi:10.1038/s12276-020-00499-2
- Lee J, Hyeon DY, Hwang D. Single-cell multiomics: technologies and data analysis methods. Exp Mol Med. 2020;52(9):1428-1442. doi:10.1038/s12276-020-0420-2
- Angelidis I, Simon LM, Fernandez IE, et al. An atlas of the aging lung mapped by single cell transcriptomics and deep tissue proteomics. Nat Commun. 2019;10(1):963. doi:10.1038/s41467-019-08831-9
- Zhu Y, Scheibinger M, Ellwanger DC, et al. Single-cell proteomics reveals changes in expression during hair-cell development. eLife. 2019;8. doi:10.7554/eLife.50777
- Stuart T, Butler A, Hoffman P, et al. Comprehensive integration of single-cell data. Cell. 2019;177(7):1888-1902.e21. doi:10.1016/j.cell.2019.05.031
- Welch JD, Kozareva V, Ferreira A, Vanderburg C, Martin C, Macosko EZ. Single-cell multi-omic integration compares and contrasts features of brain cell identity. Cell. 2019;177(7):1873-1887.e17. doi:10.1016/j.cell.2019.05.006 57. Erfanian N, Heydari AA, Iañez P, et al. Deep learning applications in single-cell omics data analysis. bioRxiv. Published online January 1, 2021:2021.11.26.470166. doi:10.1101/2021.11.26.470166
原文地址:https://zhuanlan.zhihu.com/p/465669123 |
|

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 






 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 发表于 2025-4-16 10:20
发表于 2025-4-16 10:20



