用户名
UID
Email
密码
记住
立即注册
找回密码
只需一步,快速开始
微信扫一扫,快速登录
开启辅助访问
快捷导航
门户
Portal
社区
BBS
资讯
会议
市场
产品
问答
数据
专题
帮助
签到
每日签到
企业联盟
人才基地
独立实验室
产业园区
投资机构
检验科
招标动态
供给发布
同行交流
悬赏任务
共享资源
VIP资源
百科词条
互动话题
导读
动态
广播
淘贴
法规政策
市场营销
创业投资
会议信息
企业新闻
新品介绍
体系交流
注册交流
临床交流
同行交流
技术杂谈
检验杂谈
今日桔说
共享资源
VIP专区
企业联盟
投资机构
产业园区
业务合作
投稿通道
升级会员
联系我们
搜索
搜索
本版
文章
帖子
用户
小桔灯网
»
社区
›
D、仪器区
›
分子仪器
›
生信必备技能——手把手教你做分子对接 ...
图文播报
2026庆【网站十三周
2025庆【网站十二周
2024庆中秋、迎国庆
2024庆【网站十一周
2023庆【网站十周年
2022庆【网站九周年
返回列表
查看:
10261
|
回复:
0
[分享]
生信必备技能——手把手教你做分子对接
[复制链接]
007
007
当前离线
金桔
金币
威望
贡献
回帖
0
精华
在线时间
小时
发表于 2025-1-27 13:00
|
显示全部楼层
|
阅读模式
登陆有奖并可浏览互动!
您需要
登录
才可以下载或查看,没有账号?
立即注册
×
分子对接是一种成熟的基于计算机结构的方法,广泛应用于药物发现。对接能够鉴定具有治疗价值的新型化合物,在分子水平上预测配体-靶标相互作用。
分子对接技术(Molecular Docking Method, MDM)是指通过计算机模拟将小分子(配体)放置于大分子靶标(受体)的结合区域,再通过计算物理化学参数预测两者的结合力(结合亲和性)和结合方式(构象),进而找到配体与受体在其活性区域相结合时能量最低构象的方法。配体与受体结合时,彼此存在静电相互作用,氢键相互作用,范德华相互作用和疏水相互作用。
今天小编就带大家学习如何进行分子对接。
蛋白结构和小分子结构获取及处理
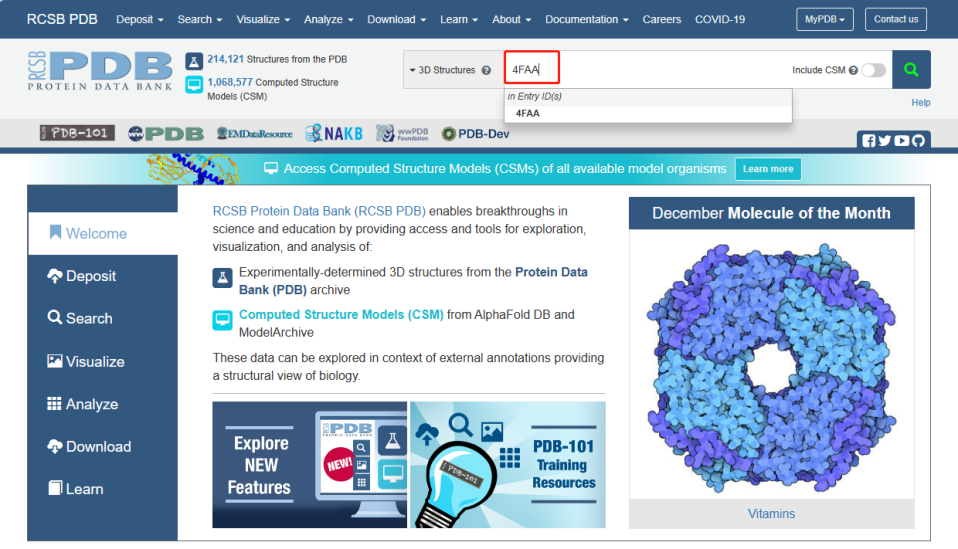
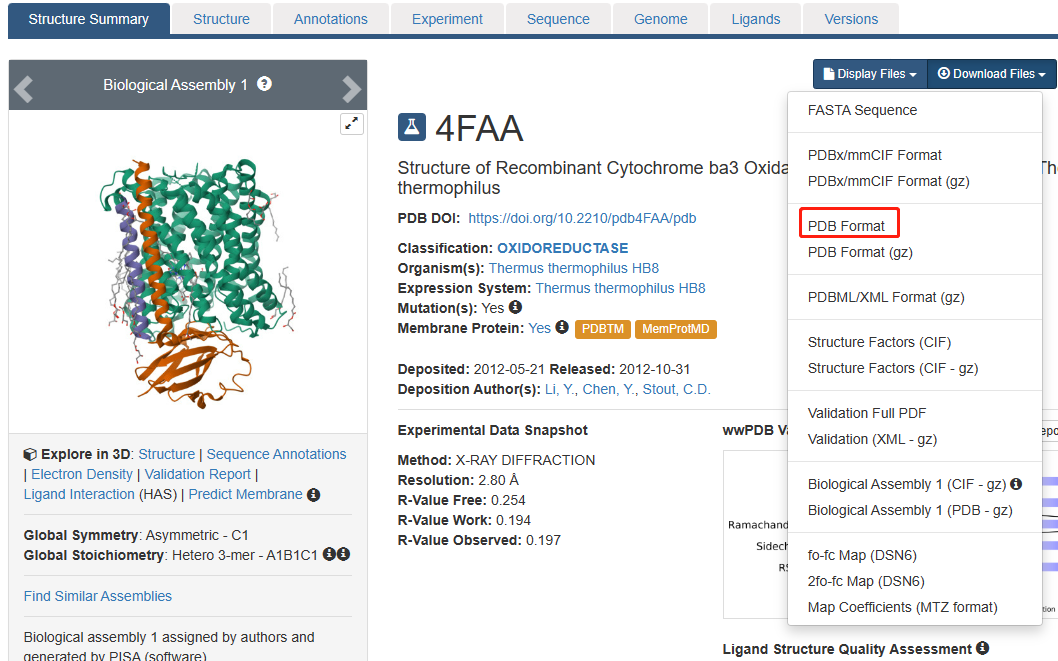
蛋白结构获取:在PDB网站(
https://www.rcsb.org/
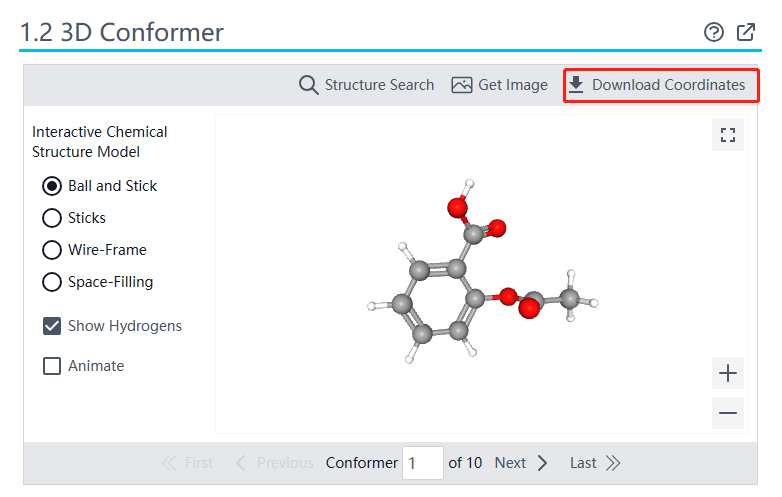
)下载蛋白的3D结构,本文以4FAA(COX2)为例。
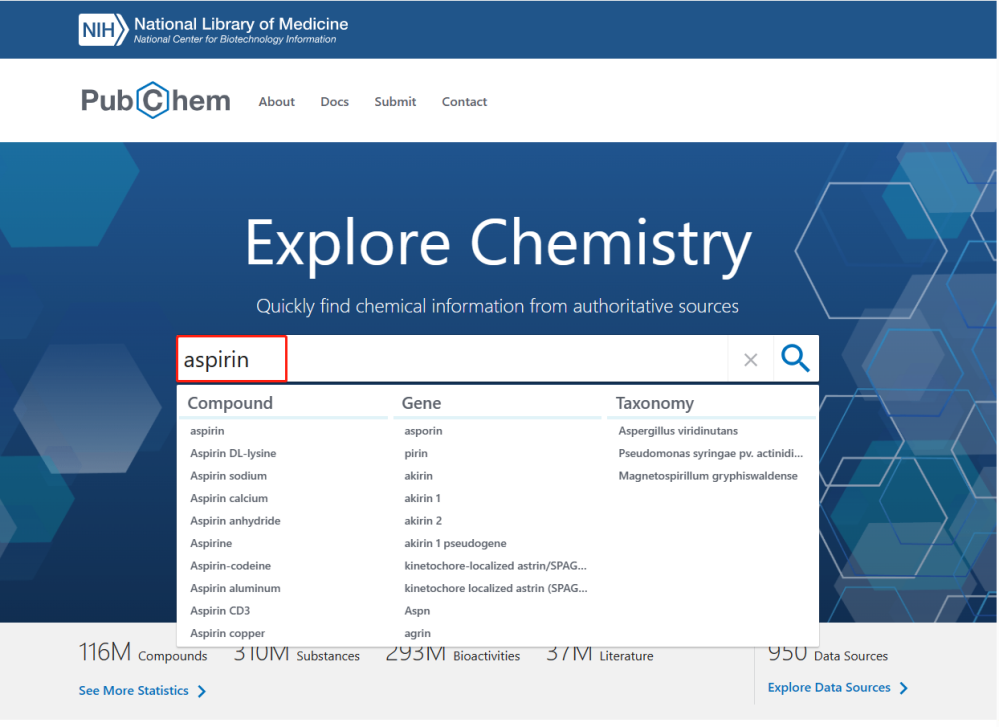
小分子结构获取:在pubchem网站(
https://pubchem.ncbi.nlm.nih.gov/
)下载小分子的3D结构(sdf格式),本文以aspirin为例。
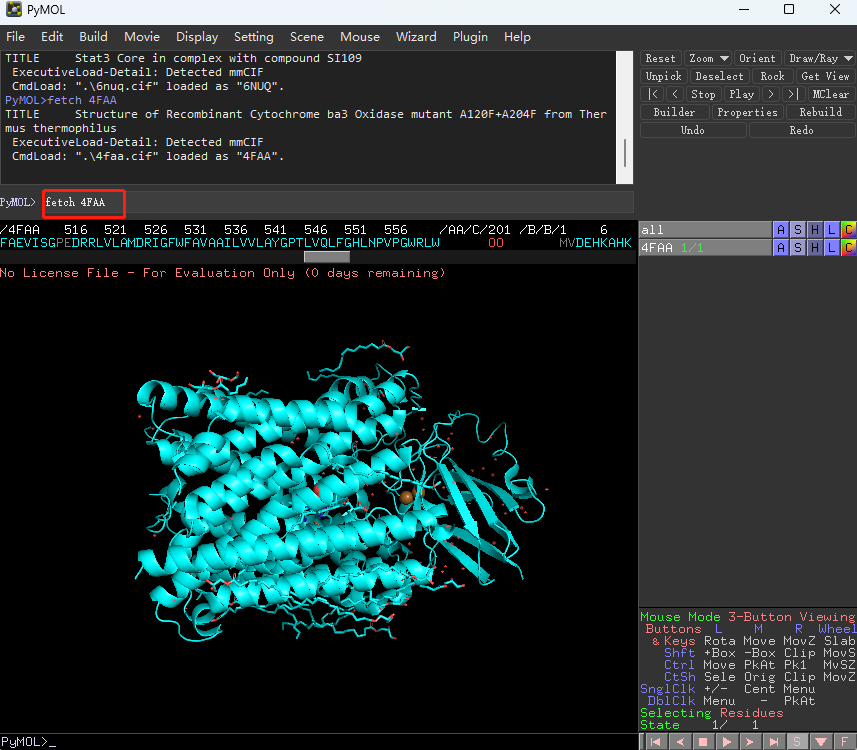
蛋白处理:打开pymol软件,fetch 4FAA:加载蛋白结构; remove solvent:去除水分子; h_add:加氢原子; 选择配体:select ligand, resn NAG;remove ligand:去除配体; iterate chain A, print(resi, resn):显示链的信息,去除副链。将结果导出为4FAA.pymol.pdb。
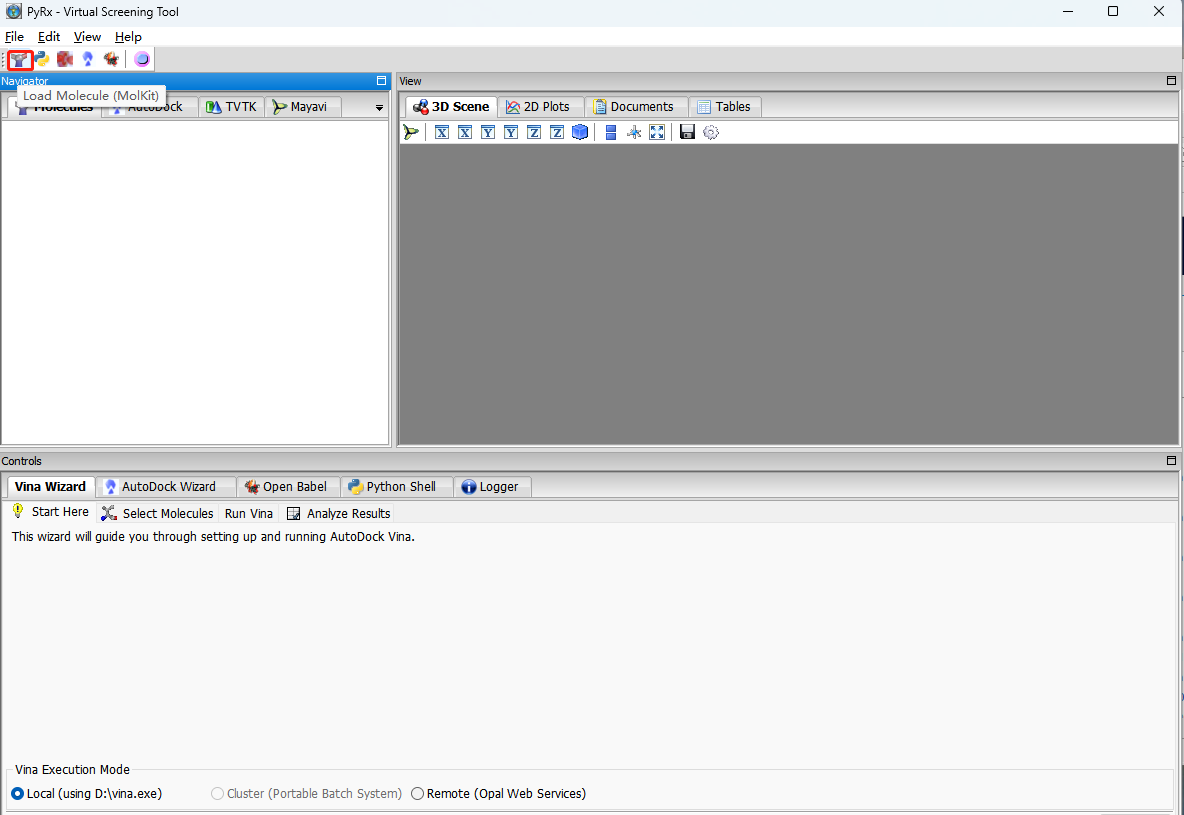
PyRx分子对接
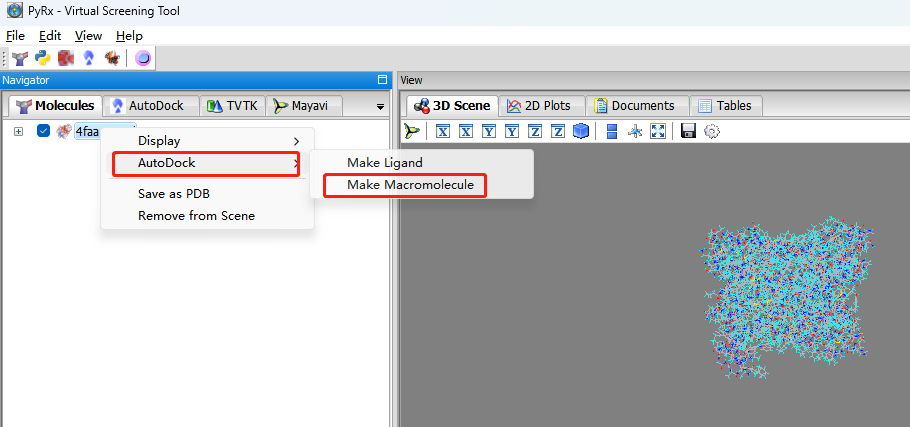
导入蛋白结构:点击File-load molecule,打开4FAA.pymol.pdb,选中蛋白,右键—AutoDock—Make Macromolecule。
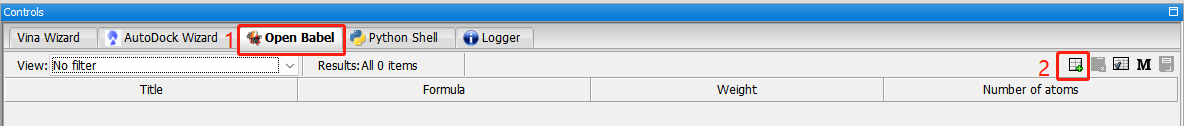
导入小分子:open Babel。
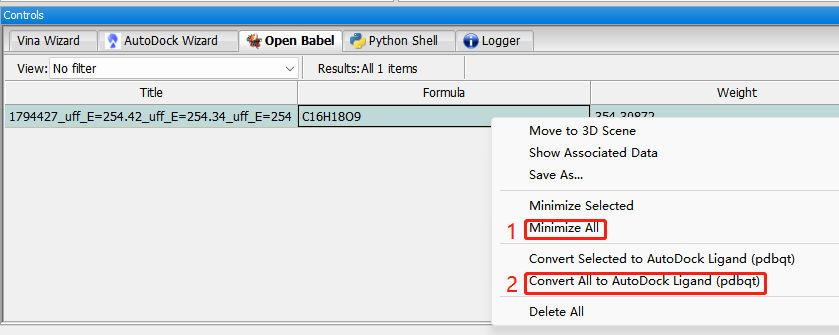
能量最小化:(1)右键—minimize all。(2)转换格式:covert all to autodock ligand (pdbqt)。
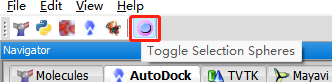
转换格式后选择配体的pdfqt文件—显示球体。
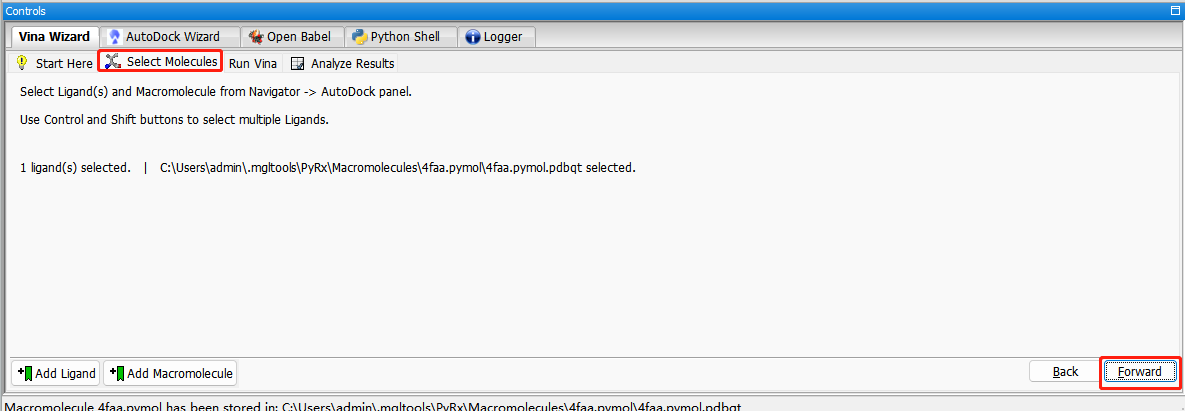
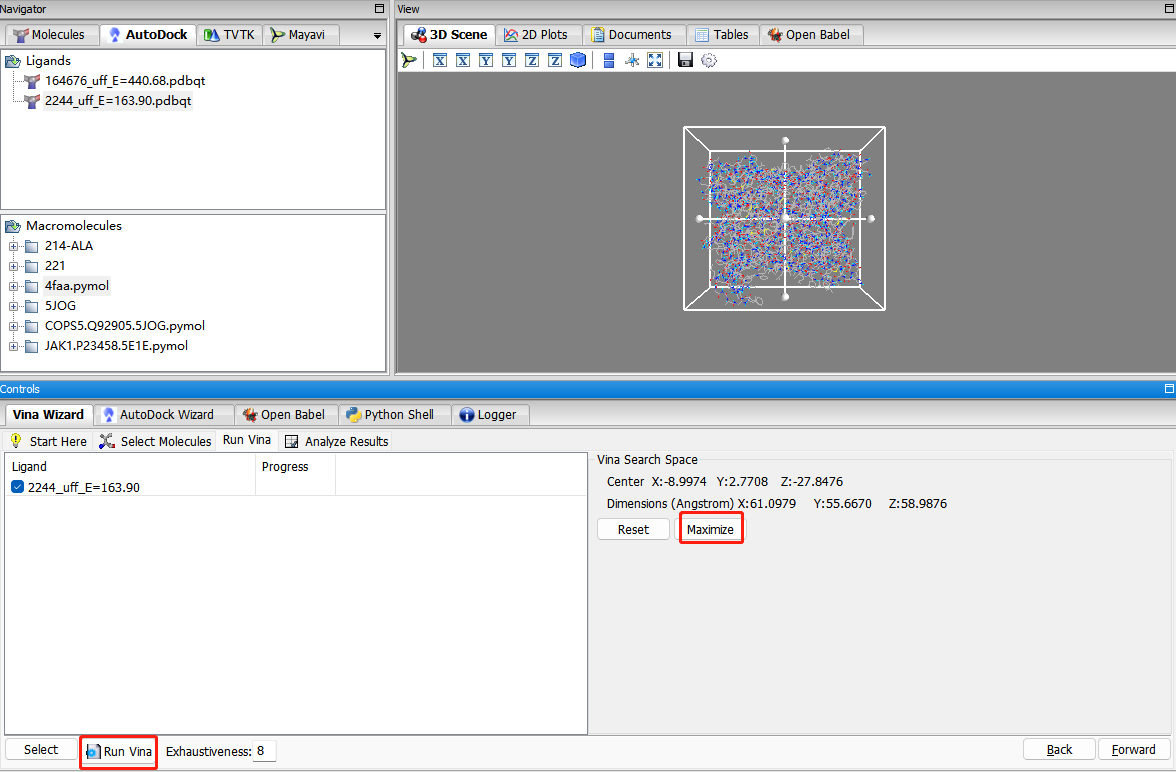
选择受体和配体分子:select molecules—forward
选择PDB靶点蛋白结合袋位置或者全选点最大化maximize。使配体完全包含在内。Run vina
对接完成后,导出表格,导出可视化文件:受体蛋白save as pdbqt格式。配体文件找到文件路径保存在受体蛋白同一文件夹中。
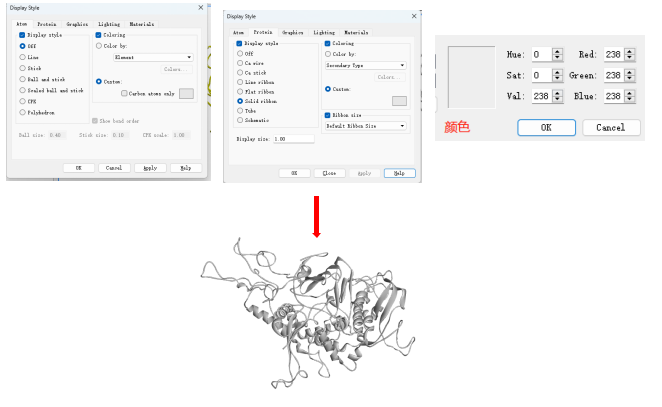
Discovery studio 处理可视化分子对接结构
打开软件Discovery studio,分别导入配体和受体的pdbqt文件。
设置颜色。选中受体结构右键 display style,选择线形及颜色。
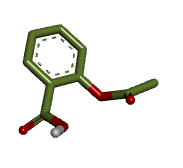
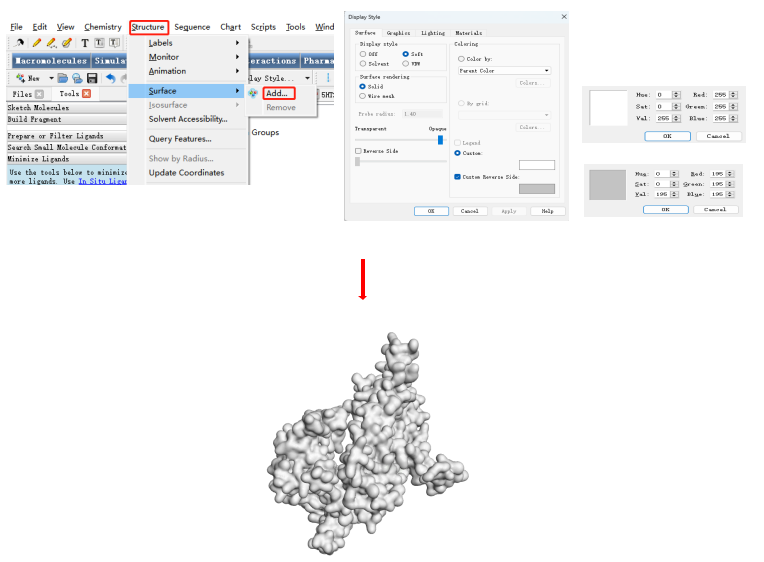
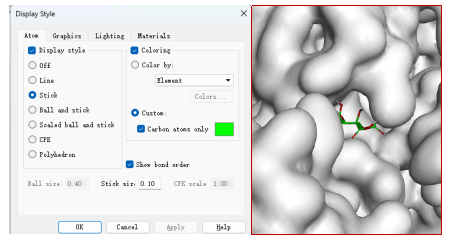
把小分子的构象复制到受体结构页面。设置配体颜色。(stick size 改变结构线条粗细)
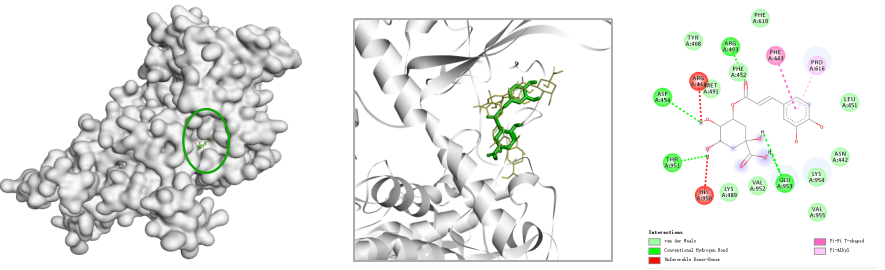
获取二维对接图像。
简单的分子对接就完成啦!Pyrx也可以进行多个小分子批量对接,大大减少了手动工作量。想要尝试的同学快行动起来吧。
原文地址:https://zhuanlan.zhihu.com/p/677834567
回复
举报
返回列表
发表回复
高级模式
B
Color
Image
Link
Quote
Code
Smilies
您需要登录后才可以回帖
登录
|
立即注册
本版积分规则
发表回复
回帖后跳转到最后一页
关闭
官方推荐
/3
AI助手<小桔子>来了!
欢迎来交流,可以回答IVD行业各类问题!
查看 »
IVD业界薪资调查(月薪/税前)
长期活动,投票后可见结果!看看咱们这个行业个人的前景如何。请热爱行业的桔友们积极参与!
查看 »
小桔灯网视频号开通了!
扫描二维码,关注视频号!
查看 »
返回顶部
快速回复
返回列表
客服中心
搜索
洽谈合作
关注微信
微信扫一扫关注本站公众号
个人中心
个人中心
登录或注册
业务合作
-
投稿通道
-
友链申请
-
手机版
-
联系我们
-
免责声明
-
返回首页
Copyright © 2008-2024
小桔灯网
(https://www.iivd.net) 版权所有 All Rights Reserved.
免责声明: 本网不承担任何由内容提供商提供的信息所引起的争议和法律责任。
Powered by
Discuz!
X5.0 技术支持:
宇翼科技
浙ICP备18026348号-2
浙公网安备33010802005999号
快速回复
返回顶部
返回列表

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 






 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 发表于 2025-1-27 13:00
发表于 2025-1-27 13:00