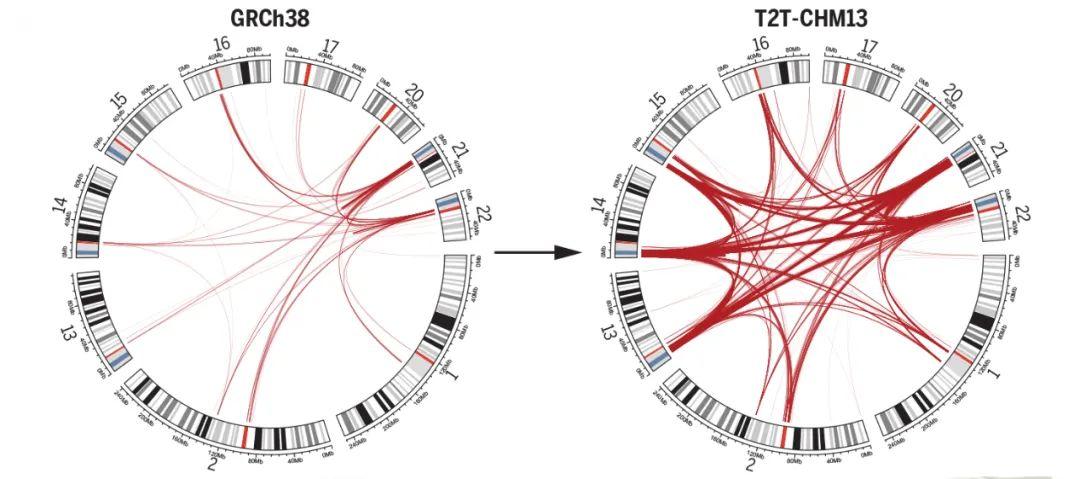
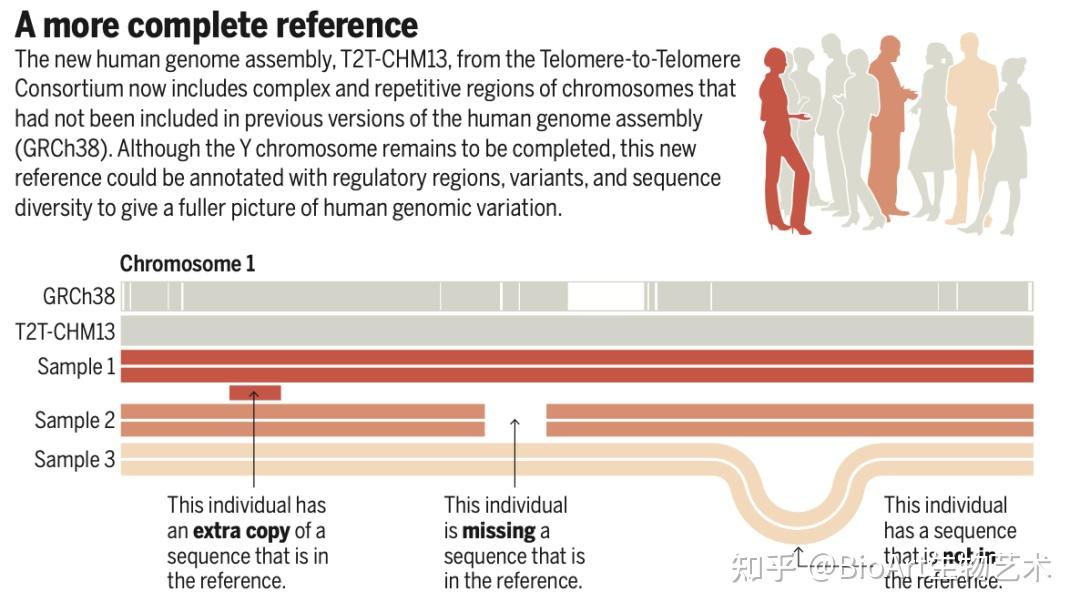
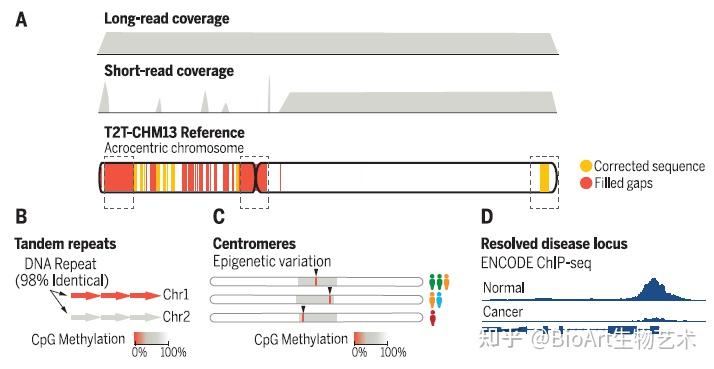
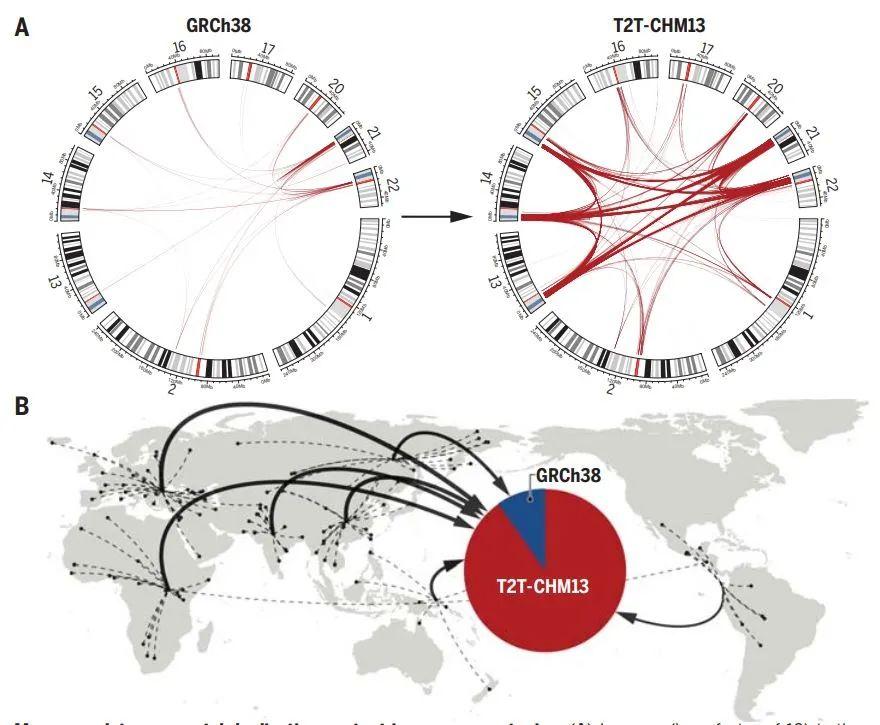
为了完成基因组的最后剩余区域,科学家们利用PacBio HiFi和Oxford Nanopore超长测序的互补方面来组装均匀纯合CHM13hTERT细胞系中的人类基因组。由此产生的T2T-CHM13参考装配弥补了这20多年来人类基因组中8%的空白,最终汇总为The complete sequence of a human genome。
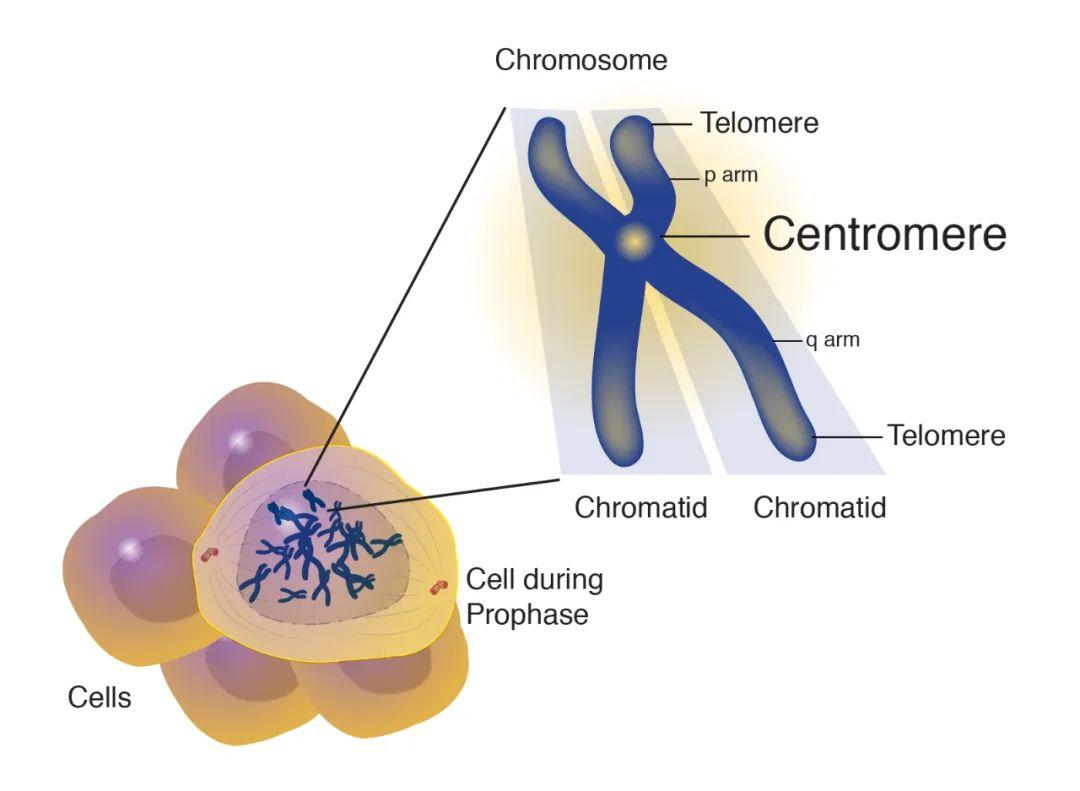
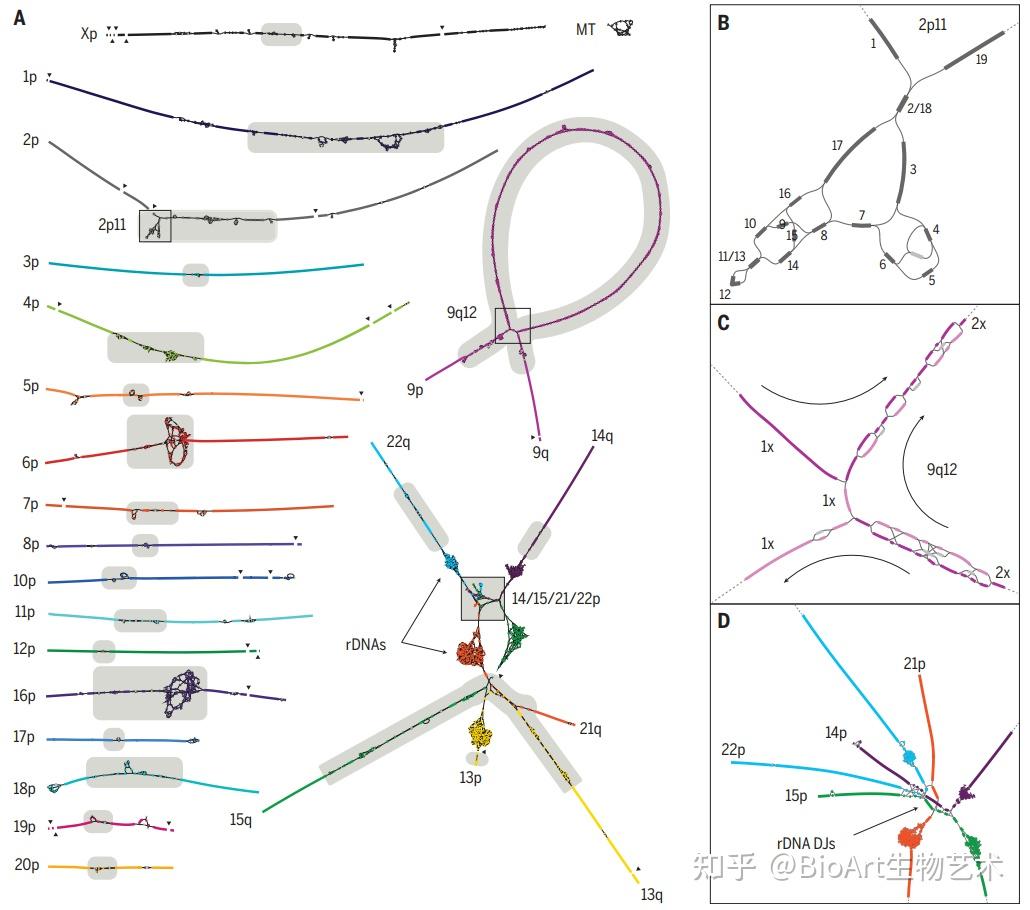
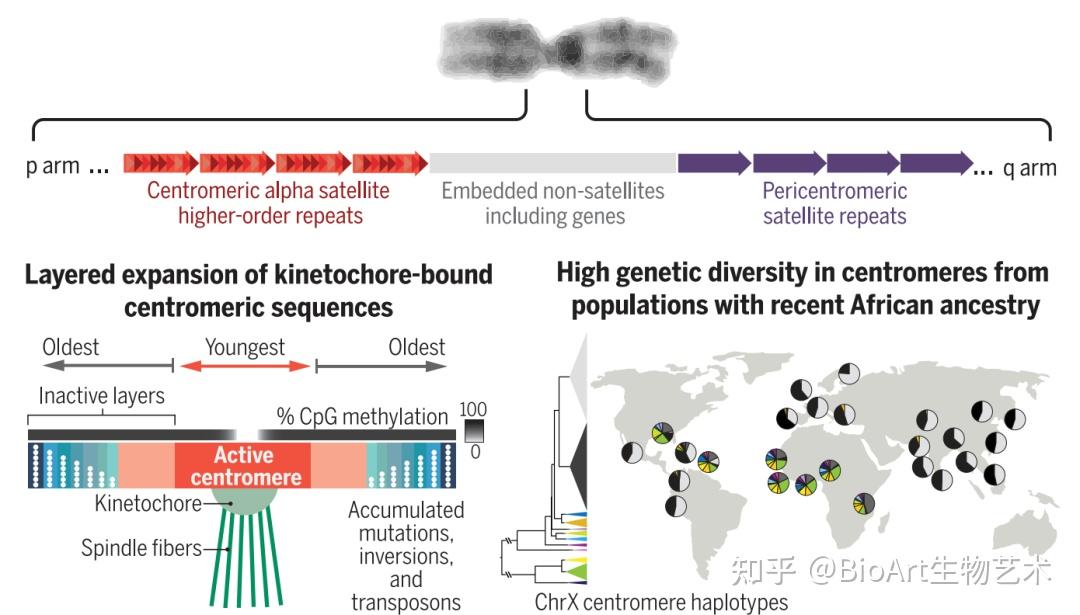
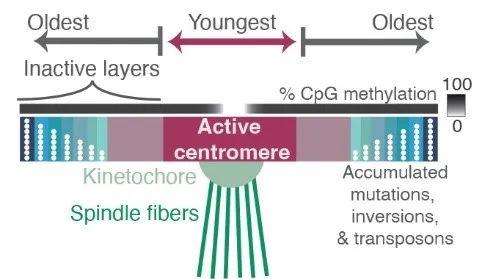
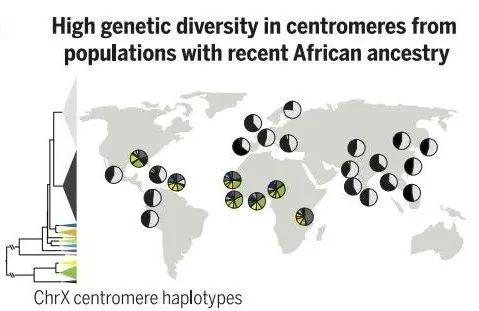
异染色质区域中的“最后一公里”难题是着丝粒。为了在细胞分裂过程中忠实地将遗传物质分配给子细胞,纺锤体纤维必须通过着丝粒的结构与DNA结合。人类着丝粒中存在大量串联重复序列,这些序列通常跨越每条染色体上的数百万个碱基对。这些重复序列的功能知之甚少,由于卫星区域的规模和重复性,以前的基因组测序工作无法生成完整的卫星区域组装,限制了研究它们的组织、变异和功能的能力。为此,端粒到端粒人类全基因组测序计划通过高精度的测序,绘制了重复区域,揭示出了这些卫星阵列不同尺度上的组织和进化模式,题为Complete genomic and epigenetic maps of human centromeres。
图4 着丝粒的前世今生
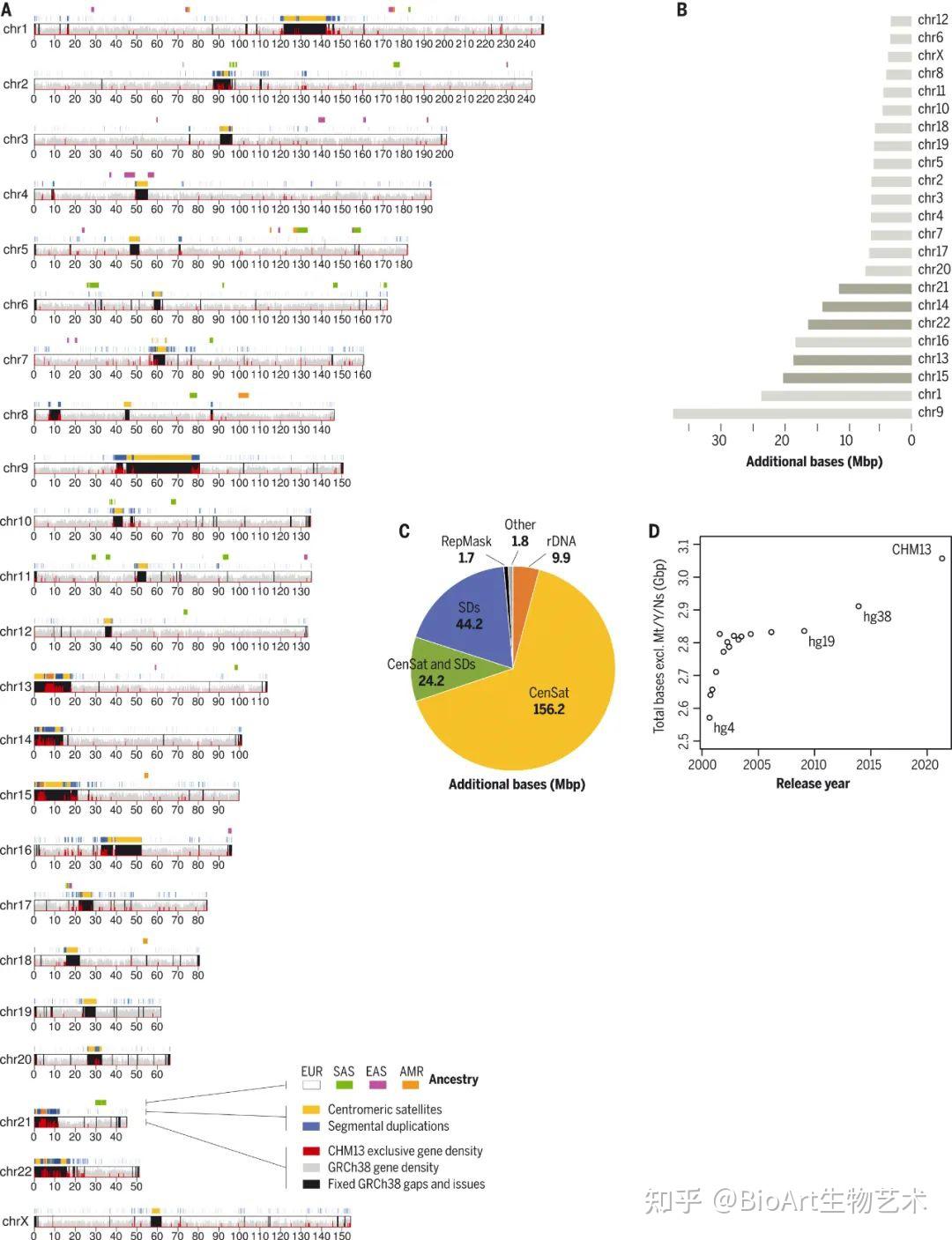
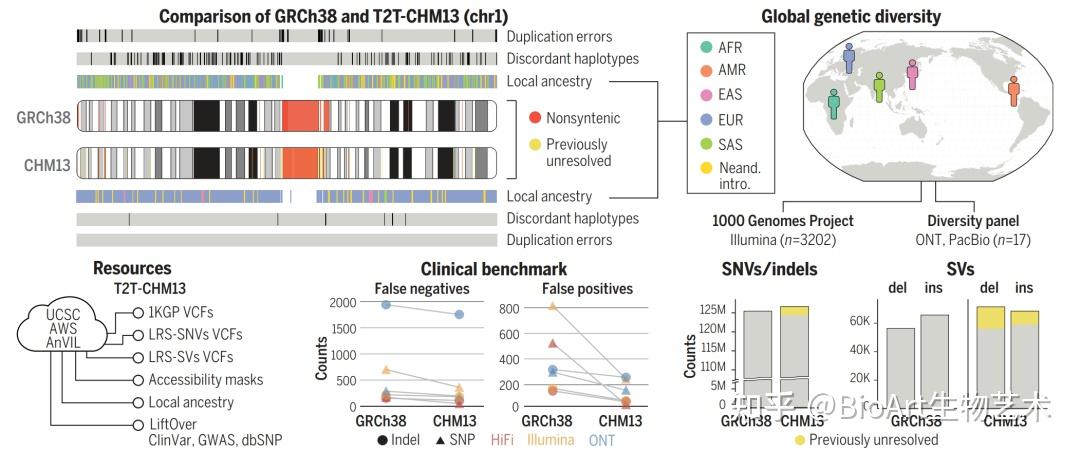
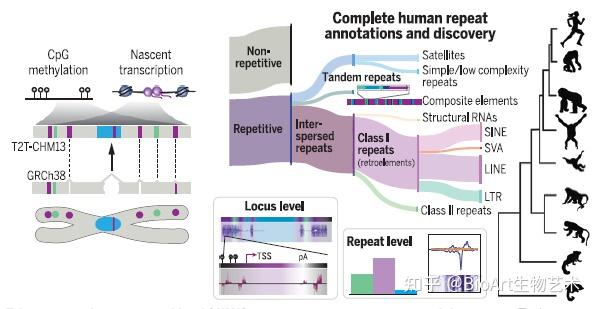
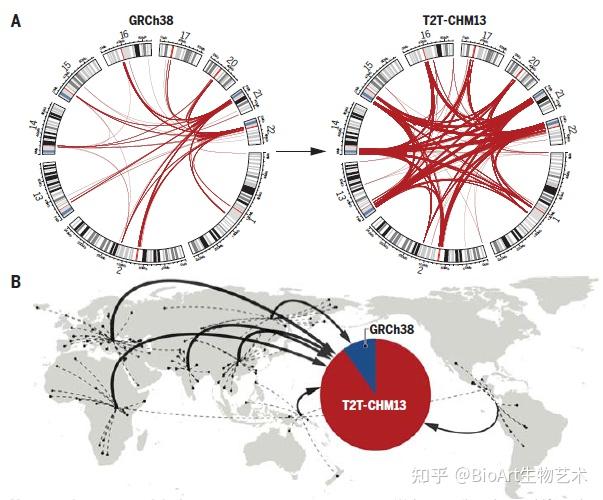
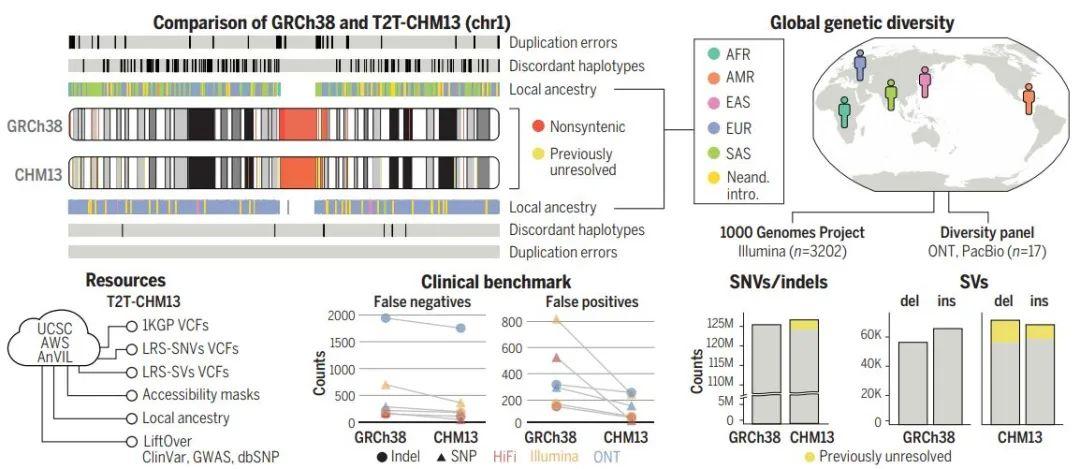
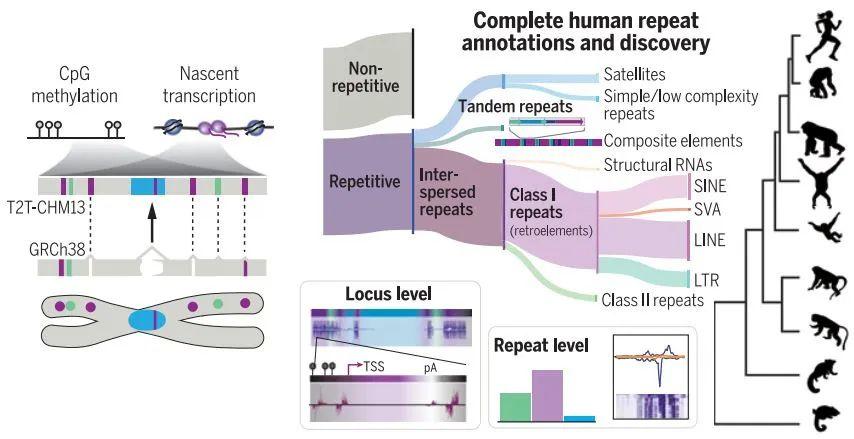
此外,还有另外三篇文章主要完善了人类表观基因组和重复序列。包括来自Johns Hopkins大学的 Winston Timp团队发表题为 Epigenetic patterns in a complete human genome 的文章;来自Connecticut大学的Rachel J. O’Neill团队发表题为From telomere to telomere: The transcriptional and epigenetic state of human repeat elements的文章;来自华盛顿大学的Evan E. Eichler团队发表题为Segmental duplications and their variation in a complete human genome。这些文章探索了以前基因组中未阐明的区域,包括近端染色体短臂、节段重复基因和人类着丝粒在内的重复序列,构建了完整的从端粒到端粒的人类基因组合集T2T-CHM13,包含了2.25亿附加的碱基对序列,全面研究基因组结构,拓宽了人类表观基因组,这为以前缺失的8%人类基因组生成完整的表观基因组。这一成就为解析这些基因组元件的作用奠定了基础。
参考资料:
[1] Sergey Nurk et al., (2022) The complete sequence of a human genome. Science. Doi: 10.1126/science.abj6987
[2] Sergey Aganezov et al., (2022) A complete reference genome improves analysis of human genetic variation. Science DOI: 10.1126/science.abl3533
[3]Mitchell R. Vollger et al., (2022) Segmental duplications and their variation in a complete human genome. Science https://doi.org/10.1126/science.abj6965
[4] Complete genomic and epigenetic maps of human centromeres. Science(2022), DOI: 0.1126/science.abl4178
[5] A. Gershman et al., Epigenetic patterns in a complete human genome. Science 376, eabj5089 (2022). DOI: 10.1126/science.abj5089
[6]S. J. Hoyt et al., (2022) From telomere to telomere: The transcriptional and epigenetic state of human repeat elements Science DOI: 10.1126/science.abk3112
[7] Complete human genome deciphered for the first time. Retrieved Apr. 1, from https://www.eurekalert.org/news-releases/946948▎药明康德内容团队编辑 本文来自药明康德内容团队,欢迎转发,谢绝转载到其他平台。 免责声明:药明康德内容团队专注介绍全球生物医药健康研究进展。本文仅作信息交流之目的,文中观点不代表药明康德立场,亦不代表药明康德支持或反对文中观点。本文也不是治疗方案推荐。如需获得治疗方案指导,请前往正规医院就诊。
⤵️喜欢我们的内容,欢迎关注@药明康德!或者点赞、评论、分享给其他读者吧!

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 






 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 发表于 2024-9-1 06:57
发表于 2024-9-1 06:57