许多研究和商业活动部门对合成 DNA 的需求不断增加。如果能够大规模并低成本地提供 DNA,工程生物学、治疗、数据存储和纳米技术将能获得快速发展。受到下一代测序和基因编辑技术成功的刺激,DNA 合成已经成为一个新兴行业。然而,>200 bp 序列的合成仍然负担不起。为了克服这些限制并开始像读取 DNA 一样有效地编写 DNA,已经开发了替代技术,包括分子组装和克隆方法、模板非依赖性酶促合成、微阵列和滚环扩增技术。
DNA是生命的信息库。自发现以来,它已成为化学、生物学和材料科学的重要研究工具。在过去的二十年中,我们在从合成基因组生成包括活体微生物在内的生物系统方面取得了显着进步。这一成功的结果是,对 DNA 的需求不断增加,推动了新技术的发展,以提供更高纯度、更高数量和更低成本的 DNA。这些要求已将商业重点转向提供合成 DNA,而不是从天然来源分离 DNA。 逐个核苷酸顺序合成多核苷酸的能力允许控制 DNA 的组成和大小。合成 DNA 序列为研究人员提供了一种多功能工具来探测生命系统,而不是依赖于从生物体中分离出的自然序列。此外,对于某些应用,例如扩增无法访问的序列,合成 DNA 便成为唯一可行的选择。DNA 合成技术的发展也可能与材料科学和纳米技术相关,例如,在 DNA 折纸中,使用非天然核苷酸或非天然骨架(例如异种核酸xeno nucleic acids(XNAs))创建新型 DNA 结构和功能。 同样,同聚多核苷酸、共同嵌段和任意多核苷酸的合成不断发展,比如在单链 DNA 作为纳米级设备或基因组工程的支架或供体材料的应用中。 下一代测序 (NGS) 的创新改进了 DNA 的读取和编辑并彻底改变了细胞和群体基因组分析,这些分析现在已应用于“大型基因组”计划。DNA可以被大规模和低成本地进行分析。然而,缺乏大规模的 DNA 合成仍然是技术进步和基因组结构和细胞功能的大规模分析的障碍。这一障碍凸显了发达的读取 DNA、识别和测序基因组的能力与欠发达的编写 DNA 以及合成和产生无限长度和复杂性的 DNA 序列的能力之间存在的差距。 在当前 DNA 合成商业化的环境下,企业要么提供他们自己合成的 DNA,要么提供即用型自动合成仪器,供研究人员在自己的实验室中制造 DNA。这两种途径都使那些缺乏专业合成技能并因此缺乏 DNA 合成“低技能”的最终用户可以进行 DNA 合成。然而,通过去技能化广泛获取合成 DNA 可能会导至合成 DNA 的滥用,这就需要进行监管以减轻滥用造成的潜在危害。 在这里,我们回顾了现有的和新兴的 DNA 合成技术,重点是工业上开发的方法,作为加速长链合成 DNA 供应的一种手段。我们还讨论了 DNA 合成为商业化带来的挑战和机遇。
为什么选择 DNA——经济驱动力 工程生物学有望为全球资源可持续性挑战提供解决方案。工程生物学的进步已经通过建立有效的合作伙伴网络和投资于自动化来满足行业需求。事实上,全球经济中所有制造业投入的 60% 可以通过生物方式生产,而 30% 的研发支出用于与生物相关的行业。DNA 合成在这方面是不可或缺的,因为它提供了工程生物学的本质——具有所需组成、复杂度和长度的 DNA 分子。 DNA技术的四次浪潮 继 2000 年代初人类基因组测序(第一波 DNA 技术浪潮)之后,“读取 DNA”的能力以超摩尔定律的速度发展,该定律预测晶体管的数量每 2 年翻一番(图 1a)。随着该领域的成熟,第二波浪潮由 DNA 从头合成和 CRISPR 基因编辑等新技术推动,这赋予了“编辑和写入”DNA的能力。这使研究人员能够通过利用读取、编辑和写入 DNA 的能力来“应用 DNA”,用于疫苗、数据存储和药物输送设备等产品或基因组工程以生成具有有用特性的生物体,例如耐热植物。DNA 应用能力的提高带来了大规模合成的需求,以应对从健康安全到环境可持续性等行业挑战。例如,COVID-19 大流行表明,疫苗需求的增长会以多快的速度压倒现有的生产能力。随着 RNA 和 DNA 疫苗继续获得批准用于治疗主要疾病,包括 COVID-19,对大规模生产DNA 的需求正在增长。同样,合成 DNA 可用于能够适应气候变化、缓解粮食安全挑战的植物。活化的 DNA 修复提高了植物对热的耐受性,同时引入合成基因使作物能够在不同条件下更有效地收集养分和水分。因此,有效的 DNA 合成对于缩小 DNA 读写能力之间的差距至关重要。 图 1:DNA 合成的技术更新。
a,以每人每天的核苷酸数量估算的 DNA 读取和 DNA 写入(合成)生产力。灰色箭头表示当前读取 DNA 和写入 DNA 之间的生产力差距。椭圆形虚线突出显示了 DNA 合成行业实现大多数重要里程碑以缩小差距的时间范围。DNA 合成数据(红线)仅适用于基于柱子的合成仪器。图中显示了每个芯片的晶体管数量(摩尔定律)以供比较。该图使用文献中可用的数据。b,报告中讨论的DNA合成技术里程碑时间表。为简单起见,并未显示所有里程碑。NTP,核苷 5'-三磷酸;PCA,聚合酶循环组装;TdT,末端脱氧核苷酸转移酶;TiEOS,不依赖于模板的酶促寡核苷酸合成。 自从首次了解 DNA 的结构(方框1)以来,已经取得了实质性的里程碑,为新产业铺平了道路。四十多年来,人们采取了简短但细致的步骤来建立用于逐个核苷酸逐步合成 DNA 的基础化学(图 1b)。开发了化学方法以可靠地提供短的 <200 个核苷酸的 DNA 链,称为寡核苷酸。这些方法针对自动合成仪进行了优化,成为基因工程和测序不可或缺的工具。在此之后,已经实现了酶促和混合方法的开发,以生成比寡核苷酸更长、更复杂的 DNA(图 1b )。公司已经将这些方法商业化,提供从定制合成到台式 DNA 打印机的服务,使非专家用户也可以进行 DNA 合成。这恰逢 DNA 应用方式的明显增加,同时暴露了 DNA 写入能力的差距(图 1a)。近年来,已经产生了数千个核苷酸长度的 DNA,突显出这种读写 DNA 能力之间的差距可能会在不久的将来缩小(图 1b)。 方框 1 DNA 的化学结构
DNA 是由四个结构单元组成的单分散聚合物,每个结构单元都包含一个碱基和一个糖残基,即脱氧核糖。每个块(腺苷 (A)、胸苷 (T)、鸟苷 (G)、胞苷 (C))与磷酸盐偶联,构成核苷酸(虚线框突出显示的胸苷)。通过磷酸二酯键连接的核苷酸形成多核苷酸链。该聚合物中的碱基通过 G 和 C 之间的三个氢键以及 A 和 T之间的两个氢键配对。这种称为 Watson–Crick 碱基配对的选择性能够形成双螺旋,其中两条互补链彼此反平行并与沿螺旋轴堆叠的碱基对(A–T、C–G)交织在一起。由于其自我互补性,双螺旋可以从两条链中的每一条链中自我复制。箭头表示不对称端的 5'-3' 方向。 DNA合成产业格局 DNA 合成行业正在迅速发展,明显转向更环保的解决方案,以减少对化学试剂和有机溶剂的依赖,这些化学试剂和有机溶剂可能对环境产生不利影响,从而减轻其昂贵的处置途径。新的行业合作伙伴关系已经形成,以在酶促 DNA 合成领域引入创新技术。这可以通过 Codexis 和 Molecular Assemblies 之间以及 Integrated DNA Technologies 和 Danaher 之间的合资企业来举例说明,这些合资企业旨在提高酶促 DNA 合成能力。有前途的技术包括质粒模板方法,例如滚动环状扩增、基因组装方法,例如 Gibson 组装或聚合酶循环组装 (PCA) ,以及不依赖于模板的酶促寡核苷酸合成 (TiEOS),它利用末端脱氧核苷酸转移酶 (TdT)作为DNA合成工具。 DNA 合成的一个关键挑战是生成 >300 个核苷酸的 DNA,这受到延伸循环效率的限制,即每个核苷酸掺入序列的效率。例如,在延伸循环效率为 99% 的情况下,包含 120 个核苷酸的寡核苷酸的理论产量约为 30% (0.99 120 × 100%)。然而,对于 200 bp 的聚合物/寡核苷酸,这减少到仅 13%。克服这个问题的尝试集中在提高 DNA 组装过程的准确性和速度上。对于商业技术,当需要 >1 kb 的序列时,化学产生的 >300 bp 多核苷酸被用作更大链的构建块。一些供应商开发的 DNA 打印机,例如 DNA Script,已经能够并行合成多个序列,这些序列可以连接在一起以产生更长的链。Molecular Assemblies 等其他公司专注于改进可能由 DNA 打印机开发商或合成基因供应商实施的合成方法。 其他供应商,如 ANSA Biotechnologies 和 Camena Bioscience,分析他们生产的 DNA 的质量,以消除用户进行进一步测序或克隆的需要。这也允许进行更多监督以应对潜在的生物安全风险。由于合成 DNA 涉及基因工程,因此存在将其用于病原体生产的风险,因此它受到监督或监管系统的约束。同样,制造 DNA 打印机的公司使用基于云的软件,可以对桌面生产模式进行一定程度的监督。表 1提供了 DNA 合成公司的示例,重点介绍了其核心技术的优缺点。 表 1 将酶促和混合方法应用于 DNA 合成的公司示例
3'-OH,脱氧核糖环的3'-羟基;C、胞嘧啶;dbDNA,狗骨DNA;G,鸟苷;NTP,核苷 5'-三磷酸;POS,亚磷酰胺寡核苷酸合成;TdT,末端脱氧核苷酸转移酶;TiEOS,不依赖于模板的酶促寡核苷酸合成。a长度取决于序列组成和复杂性。b给定技术对可想象长度的保守估计,POS 和 TiEOS 分别限制在 <0.3 和 <3 kb。c为简单起见,每种情况下给出的示例很少,这些示例对于使用类似基础技术的不同供应商可能是通用的。 制造 DNA:支撑技术 在过去的几十年里,人们对 DNA 合成技术的发展产生了浓厚的兴趣。从化学合成的二核苷酸开始,从头合成 DNA 成为可能,并在破译遗传密码的过程中得到利用。固相合成的进步激发了进一步的合成改进,这导至了 1980 年代用于 DNA 合成的亚磷酰胺化学的突破性发展,从而引入了亚磷酰胺寡核苷酸合成 (POS) 。 亚磷酰胺合成 图2A给出了一种典型的寡核苷酸固相合成过程,使用亚磷酰胺化学逐个核苷酸地构建序列 。该方法涉及逐步添加构建单元,其衍生自 5'-保护的二甲氧基三苯甲基 (DMT) 核苷酸亚磷酰胺 4。Applied Biosystems 在 1980 年代使用这种方法开发了第一台自动化 DNA 合成仪,提高了合成寡核苷酸的可及性。最初的固相方法使用塑料或玻璃固体支持物,在其上化学组装单个寡核苷酸序列(步骤c – i)。从那时起,已经使用包含多个反应位点的不同微阵列格式实现了寡核苷酸的平行原位合成,其中一个序列被组装到一个位点上,可以独立于其他位点进行控制,从而提供位点特异性的多个序列合成方式。 图 2:固相寡核苷酸合成和脱嘌呤的机制。
A,亚磷酰胺固相寡核苷酸合成的示意图。该方法在四唑激活后依次添加 5'-二甲氧基三苯甲基保护的核苷亚磷酰胺4以确保序列特异性链伸长。该过程的步骤包括合成循环,包括:质子化(步骤a)、去三苯甲基化(步骤b)、四唑活化和偶联(步骤c)、未反应的核苷酸在树脂上的加帽(步骤d)、氧化(步骤e)、脱三苯甲基化(步骤f),重复n次(步骤 g),然后从支持物上切割(步骤h)和脱保护(步骤i)以产生所需的 DNA 10。B ,作为化学合成中常见副反应的酸催化脱嘌呤的机理,包括质子化(步骤 j )、脱嘌呤(步骤k )、水解(步骤l)和消除(步骤 m)。由于嘌呤碱基(例如,腺嘌呤13)的丢失,导至无嘌呤位点14在去除碱基保护基 (PG) 所需的基本后处理步骤中很容易水解(步骤c和d )。 目前的技术使用硅作为固体支持物,可以在其上同时写入一百万个独特的寡核苷酸。与单序列合成方法相比,在硅芯片上制造的微观反应簇减少了反应体积并显着增加了 DNA 的产量,而热控制提供了一种监测每个核苷酸掺入的方法,以实现位点特异性 DNA 合成。 然而,DNA 合成的亚磷酰胺法也有缺点,包括亚磷酰胺工作台稳定性差、需要使用大量有机溶剂以及无法合成多重复序列。此外,去除 5'-DMT 保护基团 (PG) 所需的酸可以催化脱嘌呤(图 2B中的步骤j–m),这是一种有害的副反应,会导至嘌呤碱基 (A, G) 13从合成的 DNA 链丢失12,使该DNA链易受水解14和15以及过早释放16和18 。结果,脱嘌呤降低了所需寡核苷酸的产量和纯度。 寡核苷酸制造、加工和纯化的工作流程是劳动密集型的,并且在很大程度上仍然是服务提供商的领域。因此,合成能力已集中在专业试剂制造商中。Agilent Technologies、GenScript、Integrated DNA Technologies、ThermoFisher、TriLink、Dharmacon、Twist Bioscience 等领先供应商按需生产各种格式的定制 DNA(和 RNA)。对于那些希望缩短此类服务交付时间的用户,可以购买一系列仪器,例如 Cytiva 的 ÄKTA 寡核苷酸合成仪,这些仪器可以购买并日常操作。 传统上,分子生物学依赖于短的 DNA 序列,例如用于 PCR 的引物或用于分子检测、扩增和修饰应用的探针。最近,不对称 PCR 方法在扩增长度为数千个核苷酸的单个 DNA 链方面取得了进展。现在,研究人员正在寻找具有不同组成的更长序列,包括具有单碱基精度的整个基因组,这些序列必须从头开始组装。如此长的序列与亚磷酰胺法不相容,亚磷酰胺法在纯 DNA 合成中的功效在超过 200-bp 寡核苷酸序列时会降低。 要合成长 DNA,必须提高延伸循环效率以提高产量,并且必须尽量减少或避免 PG 的不完全去除和脱嘌呤等副反应。必须使用替代方法在纠错阶段从较小的链组装较长的序列。酶促方法在这方面最具吸引力,而且具有可扩展性、立体特异性和环境友好性. 酶可以介导错配识别,从而实现互补链的选择性退火,通过消除对偶联试剂的需要减少每个延伸循环中的步骤数,并减少对有机溶剂的依赖。酶可以通过扩增或在从头序列的合成中促进有或没有 DNA 模板的合成。 酶促寡核苷酸合成 酶促合成使用固相合成的原理。在固体支持物上合成的短链 DNA 可以通过 DNA 聚合酶使用核苷 5'-三磷酸 (NTP)进行延伸。DNA 聚合酶使用提供碱基配对的模板 DNA 链,从而选择传入的核苷酸。这意味着尽管聚合酶可有效扩增现有的 DNA 模板,但它们无法从头生成 DNA 序列。因此,在没有模板链的情况下,需要一种替代酶来有效地延长多核苷酸链。这种聚合酶已被鉴定为 TdT,并被整合到商业 TiEOS方法中 (图3A )。 图 3:TiEOS 的机制。
A,3'-保护核苷 5'-三磷酸 (NTP) 方法的示意图。树脂珠预载有引发剂 DNA (iDNA) ,以提供用于结合末端脱氧核苷酸转移酶(TdT) 的模板,并作为合成后切割位点。然后寡核苷酸合成以 5'-3' 方向逐步进行。TdT将 NTP 20连接到生长中的寡核苷酸链的 3' 末端,每个 NTP 在 3'-OH 处用保护基 (PG) 24 – 26保护。 洗涤树脂以去除多余的试剂和连接的焦磷酸盐副产物。在 3'-PG 去阻断或脱保护后(步骤b),树脂结合的 3'-OH 亲核试剂22可用于下一个合成循环(步骤c)。通过重复 TdT 催化的 NTP(PG) 偶联(步骤a)和解封闭(步骤b)的循环来组装完整的序列。完成后,合成的寡核苷酸4被尿嘧啶 DNA 糖基化酶从固体支持物上切下(步骤d)。B , 该方法中使用的 NTP(PG) 示例 - 由 Nuclera Nucleics 、 Molecular Assemblies 使用的3'-叠氮甲基保护的 NTP 24、由 DNA Script 使用的3ʹ-ONH 2保护的NTPs 25和由Camena Bioscience提供的3ʹ-O-2- nitrobenzyl 26。C,替代(系留)保护策略的示意图。3'-未受保护的 NTP(胞苷)通过短而不稳定的接头预先固定在 TdT 活性位点28内。然后 TdT 催化该 NTP 掺入生长中的 DNA 链30(步骤a) 并在空间上防止 NTP 不受控制的聚合,直到连接子被切割(步骤b),释放寡核苷酸32。重复该循环(步骤c)直到所需的寡核苷酸33完成(步骤 d)。Asp,天冬氨酸;DTT,二硫苏糖醇;TCEP,三羧乙基膦;TiEOS,不依赖于模板的酶促寡核苷酸合成。 不依赖于模板的酶促寡核苷酸合成 TdT 以混杂的方式在 5'- 3' 方向上延长寡核苷酸,接受四种规范核苷酸中的任何一种,导至不同序列的伴随形成。一种有效的解决方案是通过“可逆终止”机制控制核苷酸的掺入(图 3A)。该机制使用在 3' 位置用合成中断“终止子”或 PG 修饰的 NTP,这确保每个反应步骤添加一个核苷酸,随后被移除才能掺入下一个所需的核苷酸 24-26。为此,TiEOS 使用预载有化学合成的单链引发剂 DNA initiator 19的树脂珠,TdT将3'-保护的 NTP 20连接到所需序列23上。在延伸循环的每个步骤中,步骤a和b(图 3A),使用洗涤步骤去除副产物和多余的试剂,并在每个循环结束时在下一个延伸之前进行 3'-PG 的解封闭循环(图 3A中的步骤b和c)。引发剂 DNA 19在其酶促不稳定的 3' 末端包含一个高度特异性的脱氧尿苷裂解位点。该位点在合成完成后被尿嘧啶DNA糖基化酶切割以从树脂释放组装的序列23 。 TdT 方法学作为 DNA 合成的主要范例 随着越来越多的公司测试这种方法,已经报道了 TdT 的几个重要和独特的局限性。首先,该酶表现出更倾向于掺入某些核苷酸。这种偏差可能会增加特定序列错误的发生率。其次,TdT 仅适用于单链 DNA。这归因于酶中的套索状环,它充当空间屏蔽,防止双链 DNA 模板进入酶的活性位点。因此,如果构建中的链开始形成二级结构,合成效率就会降低。第三,与所有 DNA 聚合酶一样,TdT 催化的磷酸转移需要二价阳离子才能从 NTP 合成 DNA。然而,与通常需要 Mg 2+来催化 DNA 分子合成的其他 DNA 聚合酶不同,TdT 可以使用各种二价金属阳离子,例如 Co 2+、Mn 2+、Zn 2+和 Mg 2+,具有由阳离子身份定制的 NTP 掺入。例如,Mg 2+的使用有利于三磷酸脱氧鸟苷和三磷酸脱氧腺苷的结合,而 Co 2+促进三磷酸脱氧胞苷和三磷酸脱氧胸苷的结合。至关重要的是,这种偏差扩展到 DNA 合成中使用的受保护 NTP ,促使研究人员开发减轻偏差的方法。TdT 的其他特征会影响 PG 的选择和合成效率,包括 DNA 磷酸化能力和酶的磷酸酶活性。因此,当生长的寡核苷酸链暴露于 NTP 混合物时,TdT 会优先掺入某些核苷酸,从而导至合成不同长度的均聚链。 为了解决这些缺点,正在探索不同的方法。与肽合成类比,可以定制微波辐射以使用作用于双链 DNA 的 DNA 聚合酶加速合成,或将所需的双链 DNA 转化为 TdT 可访问的单链形式。 为了避免 TdT 将 NTPs 随机掺入到生长的 DNA 链中,已经为 NTPs 24-26开发了合适的 3'-PG (图 3B),这促进了包括 3'-PG 去封闭、树脂洗涤和偶联的顺序合成循环NTP 的数量(图 3A)。该循环构成了技术优化的 TiEOS,已被 DNA Script 和 Nuclera Nucleics 等多家公司采用。 这种方法的重要优化涉及 3'- PG的设计,例如,DNA Script 选择 3'-ONH 2 -受保护的 NTP 25,而 Nuclera Nucleics 和 Molecular Assemblies 更喜欢叠氮甲基终止子24和 Camena Bioscience 似乎支持 2-硝基苄基26作为 3'-PG。尝试使用其他PG来保护3'-OH 和NTP 的碱基,其他平行努力侧重于 PGs 用于 XNA 合成。然而,TdT 必须能够在其活性位点容纳受保护的核苷酸,这限制了 PG 的选择或需要重新设计酶以与 3'-PG 相容。实际上,由于酶71活性位点的空间位阻,3'-PG NTP不是 TdT 的天然底物,并且它们的开发受到供应商的严密保护。TdT的重新设计可以解决这个问题,也可以帮助开发耐热的TdT 。DNA Script 、 Nuclera Nucleics和Molecular Assemblies是该领域的活跃参与者,而 Camena Bioscience 开发了专有的高保真酶组合,可实现无模板 DNA 合成(表1 )。 其他公司采用替代方法,通过开发 3'-OH 保护策略来暂时限制不断增长的寡核苷酸链。例如,Molecular Assemblies为传入的 NTP 提供阻断基团,以在空间上保护其 3'-OH 免于延伸,直至移除。在另一种策略中,ANSA Biotechnologies 通过可切割接头将 TdT 连接到传入 NTP 的碱基上,以防止形成均聚核苷酸束(图 3C)。NTP 28的 α-磷酸基团与生长中的寡核苷酸27的 3'-OH 发生反应,而其未受保护的 3'-OH 仍然受到酶的空间屏蔽30,这可以防止聚合。切割接头可释放 TdT 31和延长的寡核苷酸32。通过重复循环,步骤a–c,可以组装和释放所需的序列33。 化学合成寡核苷酸的产量、纯度和可达到的长度取决于每个偶联循环的有效完成。虽然 TiOES 中使用的两步循环是对 POS 中所需的四个步骤的改进,但 TiEOS 不太可能提供具有成本效益和时间效益的全长基因合成。接近定量的 99.9% 的延伸循环效率导至 1,000-bp(或 1 kb)DNA 链的产量 <37%。相比之下,DNA Script 报告的 99.7% 效率将导至产量低于 5%。然而,如果是 99.9%的效率,3 kb DNA 的产量也可以实现 <5%。例如,Camena Bioscience 应用了他们专有的从头合成和基因组装技术——gSynth——构建了一个 2.7 kb 的质粒载体 pUC19。 随着合成的进行,> 3 kb 的多核苷酸链可以形成稳定的二级结构(例如发夹),对延伸循环效率产生不利影响。微波处理可能会缓解这个问题,但仍在 3 kb 范围内。 尽管存在局限性,与 POS 相比,TiEOS 通过最大限度地减少可能的杂质数量、使用“绿色”试剂以及每个合成周期依赖更少的步骤来降低粗制寡核苷酸的复杂性。与 POS 相比, TiEOS 的这些优势提高了产品纯度和质量,但仍未实现定量延伸或解决二级结构形成对 DNA 合成的不利影响。因此,TiEOS 被视为合成 <3 kb DNA 的有前途的“绿色”方法。为了合成更大的结构(即基因簇或染色体),TiEOS 可用于生成较短的片段,然后可以通过 Gibson 组装或 PCA 进行连接。 无限长度DNA技术 DNA 的互补性(方框1)和大量能够聚合、切割、切口、连接和突变 DNA 的酶促成了各种组装方法的发展。通过改进酶和装配标准,提高了单步合成DNA分子的准确性和数量,已应用于最小细菌基因组合成和合成酵母染色体。在其他地方详细综述了 DNA 组装方法后,我们在此重点介绍 DNA 合成工作流程的两种基本方法,即Gibson组装和PCA 。 吉布森组装 Gibson 组装是一种酶方法,用于补充 POS 和 TiEOS 方法。尽管这种方法对于短链(<100 个核苷酸)的合成效率低下,但它可用于组装大的 DNA 片段 (图4A )。吉布森组装从两个 DNA 双链体34和35开始,它们具有互补的末端重叠区域。这些 DNA 双链体的每条链都被 5'-末端的核酸外切酶降解,产生双链体36和37的 3'-“粘性”末端。然后将这两个双链体的粘端逐步退火b并由聚合酶修复,聚合酶使用碱基配对相互作用将缺失的核苷酸添加到两条链中。然后 DNA 连接酶将每条链的核苷酸缝合在一起,形成所需的双链体产物38。 图 4:DNA 合成组装方法的示意图。
A、吉布森组装。选择具有互补末端重叠区域(黑色)的两条双链 DNA 链34和35。用 T5 核酸外切酶消化(步骤a)在 5' 到 3' 方向降解 DNA 双链体的每条链,产生36和37的粘性末端,然后两个 DNA 双链体之间的互补粘性末端退火 (步骤b)。然后结合 Phusion 聚合酶和 Taq 连接酶(步骤 c)将两个短 DNA 双链体连接成一个单一的长 DNA 双链体构建体38。重复该过程(步骤d)以进行基因组装。 B , 聚合酶循环组装 (PCA)。设计高纯度合成寡核苷酸39 ,使得互补重叠的退火产生所需的长双链 DNA 构建体(步骤a)。然后使用 DNA 聚合酶(步骤b)从43中的一个步骤或39中的两个步骤中组装所需的构建体以产生模板40。41的PCR 扩增(步骤c和d)扩增所需的长双链 DNA 结构42。 多轮 Gibson 组装产生大量基因片段,可用于一系列应用,例如从蛋白质表达到转录控制。然而,这个过程仍然很费力。非自动化基因组装非常耗时,而且需要大量的高纯度寡核苷酸。寡核苷酸纯度对于确保正确组装也很关键,即使是很小百分比的缺失也会在所需 DNA 的开放阅读框中产生大量移码突变 - 由酶转录成 RNA 的 DNA 部分。即使是单个缺失也会改变阅读框,损害 RNA 转录,从而使 DNA 无法使用。因此,最终的基因产物被克隆到质粒中并转化到细菌菌株中以确认所需 DNA 序列的存在。较长基因的合成通常需要多次克隆和重复的 Gibson 组装步骤,从而导至额外的成本和较长的交付周期。 聚合酶循环组装 由于 PCR 的发展,DNA 的扩增和测序现已成为常规。Watson–Crick 碱基配对与 PCR 结合使用,开发了一种称为 PCA 的技术中将合成寡核苷酸池拼接在一起的方法。在 PCA 中,被称为“有义sense”的目标寡核苷酸通过互补突出端与对应于互补反义链的寡核苷酸退火(图 4B )。每个寡核苷酸,除了那些位于每条链的 5' 末端的寡核苷酸外,都与相反链中的两个互补寡核苷酸杂交。这产生退火构造39和43,在有义链和反义链中存在交替的“缺口”。然后使用聚合酶填充间隙以生成用于 PCR 扩增的双链 DNA 模板40和41。在这个装配阶段之后,引入与双链 DNA 模板的 5' 末端互补的外部引物以进行 PCR 反应,从而扩增目标序列以产生最终产物42。使用这种方法,已经从短的、化学合成的寡核苷酸产生了 >2.5 kb 的质粒。 与 Gibson 组装一样,PCA 的性能可能会受到合成寡核苷酸杂质的影响。PCA 的其他缺点包括该方法依赖于单个克隆的序列确认,以及依赖高保真校对 PCR 酶,这些酶必须用于复制构建的基因以防止扩增过程中发生突变。然而,由于迭代合成多核苷酸长度的限制,Gibson 组装和 PCA 仍然是制造大 DNA 的主要实用选择。 新兴的商业化技术 开发制造 DNA 新方法的公司专注于满足两个主要要求:更长的 DNA 序列或更多数量的并行 DNA 构建体。模板化和独立于模板的方法都是为大规模生产和长 DNA 组装而开发的。越来越多的公司将重点放在 DNA 合成服务上,这仍然具有很高的竞争力,并且需要对合成 DNA 的分销进行更严格的控制。自动化提供了最大限度地减少专家参与 DNA 合成的机会,并且正在通过提供台式 DNA 打印机来实现。通常,行业会根据复杂性和长度为特定的 DNA 目标定制合成方法。这是由具有挑战性的局部应用驱动的,例如 DNA 疫苗或基因治疗的合成。这些应用证明了提供高产量和高纯度 DNA 产品的价值。随后讨论了一些令人兴奋的工业发展,以举例说明 DNA 合成领域的进展。 热控合成 Evonetix 提出的并行 DNA 合成的渐进解决方案是热控合成。该方法与亚磷酰胺和 TiEOS方法兼容,并提供 DNA 文库的合成,序列固定在硅芯片的离散热控制反应位点上。加热允许从特定反应位点的末端选择性地切割 PG 47(亚磷酰胺为 5',TiEOS 为 3',图5中的步骤a)。然后可以将整个芯片暴露于 TiEOS 或亚磷酰胺延伸循环,选择性地仅延伸固定在加热反应49上的寡核苷酸。未加热的位点保留其热不稳定的末端 PG,使这些链无法用于延伸46和48(图5)。 图 5:热控 DNA 合成的示意图。
末端保护的寡核苷酸链44和45固定在离散的反应位点(位点 1 和 2)上。所选位点(位点 2)的热加热会裂解末端保护基团 (PG) 47(步骤a ),从而能够选择性地延长该位点上的链(步骤b和c)所需寡核苷酸的延长是通过TiEOS或亚磷酰胺方法选择性地生成49。重复步骤a– c在其他选定的反应位点(例如,位点 1)上顺序生成定制寡核苷酸50 (步骤d和e)。所选位点的热辅助试剂处理会切割安全捕获接头并从所选位点释放寡核苷酸53 (步骤 f)。释放的寡核苷酸(步骤g)与互补的芯片结合寡核苷酸退火52产生完美退火的双链 DNA 54,其变性温度高于由错配寡核苷酸形成的 DNA 双链体55. 反应位点 1 的加热允许错配的寡核苷酸56被洗掉 (步骤h )。位点 1 的热辅助试剂处理切割安全捕获接头并从芯片释放所需的双链体 DNA 57 (步骤i)。该释放的 DNA 与芯片结合的互补 DNA 双链体58退火以形成带切口的构建体59(步骤j)。重复该过程以延长双链 DNA,直到装配所需的基因。 与其他 DNA 合成方法一样,延伸循环效率是限制因素。在每个反应位点,预计 PG 的热解不足百分比。在每个延伸周期中,缺失序列都会积累,从而产生与所需产物相似的杂质。Evonetix 通过一个对热辅助化学裂解不稳定的接头将每个固定化的寡核苷酸连接到芯片上,从而解决了这个问题。一旦组装了所需的链53,就将它们固定到的芯片上的位点加热,导至接头裂解并将这些链释放到溶液中。这些释放的链可以与寡核苷酸互补52,它们仍然固定在芯片上,随后可以一起退火以产生双链 DNA 分子54。任何不完全退火的寡核苷酸对55,例如,由于截短的序列,可以在比所需 DNA 54更低的温度下热变性。这种热纯化过程去除了不正确的序列56 ,产生了具有所需碱基配对57 的双链 DNA 产物。如果这些双链 DNA 对具有与固定在芯片的另一个位置的58链互补的粘性末端,然后顺序对可以退火到一个“切口”构造59。重复这个过程会产生双链产物,其长度几乎是无限的。链中存在的切口可以通过 DNA 连接酶修复成所需长度的双链 DNA,并且可以通过 PCR 扩增构建体。Evonetix 预计将提供基于该技术的桌面 DNA 打印机。这些即插即用仪器将以在云中实施的用户界面和设计算法为特色,以实现对基因合成生物安全性的控制。 文库基因合成 Ribbon Biolabs 开发了一种方便的合成长(>10 kb)双链 DNA 的方法,使用双链寡核苷酸池的会聚组装(图 6A)。该方法需要合成一个包含数万个高纯度 5'-磷酸化单链寡核苷酸和 8-26 个核苷酸长度的文库,包括 DNA 合成所需的所有构建模块。每个寡核苷酸在库中都有一个指定的 5'-磷酸化反向互补链,在 5'-末端为每条链设计了四个核苷酸的退火突出端。组装过程需要对互补寡核苷酸60和61进行变性和退火,以及62和63以生成双链 DNA 构建体64和65的文库,每个构建体在两条链的 5' 末端都有两个四核苷酸粘性末端。 图 6:基因合成策略。
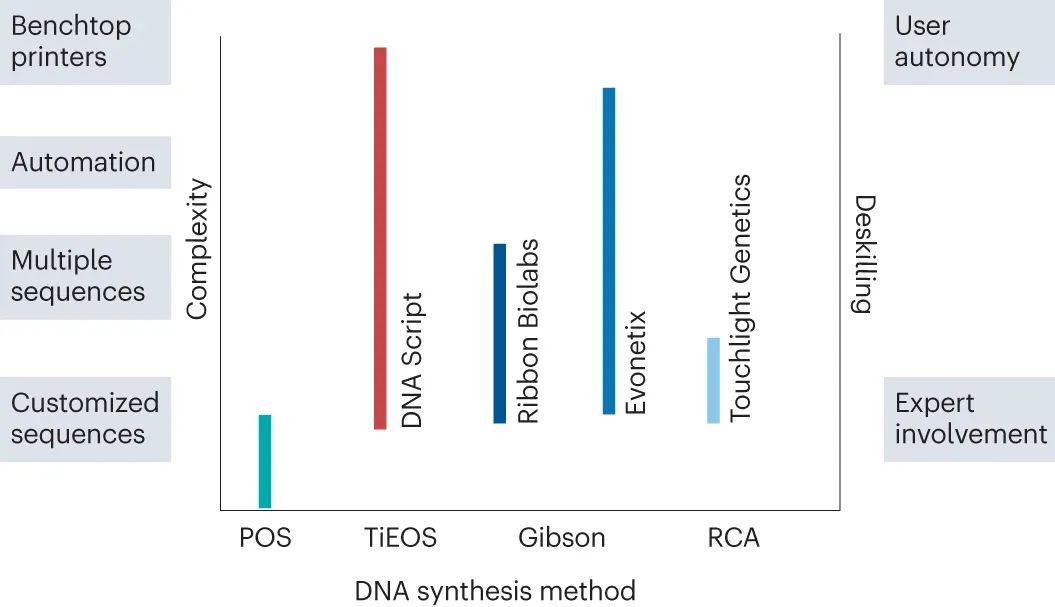
A,来自不同寡核苷酸文库的基因合成。5'-磷酸化有义寡核苷酸链60和61与互补的 5'-磷酸化反义链62和63退火(步骤a )。由此产生的 DNA 双链体64和65具有两个 5'-悬垂端或粘性末端,用于将双链体退火(步骤b)成延伸的“缺口”双链体66(“缺口”以洋红色突出显示)。然后使用 T4 DNA 连接酶(步骤c)将切口位点的寡核苷酸缝合成细长的、更大的 DNA 双链体67。重复退火和连接循环,直到组装所需基因 (步骤d)。B、DNA微阵列。在小型化芯片上生成具有 3' 末端和 5' 末端 DNA“条形码”的定制单链寡核苷酸文库68(步骤 a )。这些序列被切割(步骤b) 从微芯片中产生具有一系列 DNA“条形码”的模板寡核苷酸池(为清楚起见,仅显示黑色和棕色“条形码”两个)。引物选择性地与“棕色” 69或“黑色” 70 DNA 条形码退火,并根据条形码在其 3' 和 5' 末端的身份,通过PCR (步骤c)特异性扩增寡核苷酸。由此产生的双链 DNA 结构71和72仍然在其末端包含 DNA 条形码,必须在基因组装之前将其移除。DNA 条形码被切割(步骤d) 通过 IIS 型限制性核酸内切酶 (REN) 从双链 DNA 71和72中分离出来,产生具有粘性末端的序列73和74的组装池。双链 DNA 片段通过互补粘端退火(步骤e),并通过 Gibson 组装组装成所需基因75和76。C , 滚环放大 (RCA) 。具有所需基因盒(绿色)和原端粒酶位点(品红色)的模板质粒 DNA 77被热变性(步骤a)创建单链模板78。互补引物结合到端粒酶位点79(步骤b),模板通过 RCA 扩增(步骤c-e),产生双链多联体 DNA 82,交替拷贝所需的盒(绿色)和不需要的质粒主链(黑色)。然后,蛋白端粒酶在其识别位点切割(步骤f)双链体并连接切割末端,产生共价闭合的“doggybone”DNA(dbDNA)84和环状质粒 DNA 83作为副产品。质粒 DNA 的环状骨架随后被 REN切割(步骤g )并被外切核酸酶消化(步骤h)。 具有 5'-磷酸化、四核苷酸粘性末端的双链 DNA 片段64和65然后在步骤b中退火,并在步骤c中连接在一起以进行会聚合成,从而产生更大的双链 DNA 67以供进一步组装。这些构建块的退火和连接的重复循环给出了所需的双链结构。 末端双链 DNA 块具有单个“平末端”和单个“粘性末端”以产生线性双链 DNA 产物。一旦获得最终的 DNA 双链体,就可以使用高保真聚合酶通过 PCR 对其进行扩增,从而为客户提供产品产量。因此,Ribbon Biolabs 的这项技术类似于收敛convergent的 Gibson 组装方法。 从 DNA 微阵列合成基因 Twist Bioscience 通过在硅微阵列芯片上微型化和执行基因合成,开发了一种类似有效的方法。使用喷墨印刷生成由 25,000 个离散反应位点组成的网格。然后将专业试剂运送到每个站点。该方法能够从数万个文库中选择性地延伸几个所需序列,并提高延伸效率(图 6B)。该方法中使用的低浓度和体积(大约飞摩尔)允许大量过量使用起始试剂,而酸性 5'脱三苯甲基溶液通过碱性氧化中和以防止脱嘌呤。组装序列的产生量相对较低,这就需要使用 PCR 来产生足够的 DNA 用于基因组装。微阵列提供复杂的 DNA 库68,其中可以包括互补双链体的两条链。退火后,双链体用于通过 PCR 进行模板特异性69和70扩增,以生成大量选定序列71和72,分别与引物有效杂交。在这种形式中,双链 DNA 可以从复杂的序列池中选择性地平行扩增。然后,通过 PCR 扩增的双链 DNA 71和72的装配子池被 IIS 型限制性核酸内切酶消化,分别产生粘端双链体73和74。然后将这些双链体用作 Gibson 组装的构建块,以组装所需基因75和76,其成本仅为传统柱合成寡核苷酸的一小部分。小型化也使其他领域受益。值得注意的是,微阵列已被证明有助于优化 TiOES 平台上寡核苷酸的平行合成,包括起始链和化学修饰的 NTP 对酶促 DNA 合成的影响。微阵列还促使早期考虑 DNA 纳米制造、合成多重化以及酶与替代聚合方法的兼容性。 滚环扩增技术 Touchlight Genetics 已将一项技术商业化,该技术使用线性闭合“狗骨”DNA (dbDNA) 扩大大 DNA 的制造,dbDNA 以其类似于狗骨的结构84而被命名(图6C)。dbDNA是通过质粒模板 77 的滚环扩增产生的。模板必须设计为包含所需的表达盒,例如,反向末端重复,直接位于两个 28 核苷酸的原端粒酶识别位点之间。变性质粒模板78在存在与原端粒酶识别位点结合的引物的情况下,由 DNA 聚合酶扩增79。一旦质粒被复制,DNA 聚合酶继续通过滚环扩增重复复制质粒模板,从模板中取代任何预先存在的合成链81。然后,聚合酶与这些释放的 DNA 单链结合,并复制互补链以生成连环双链 DNA 82。添加蛋白端粒酶以产生双链 DNA 断裂并形成发夹环以重新密封末端,从而产生 dbDNA 84和环化质粒 DNA 副产物83。 限制性核酸内切酶经过仔细选择,以便它们可以消化不需要的质粒主链83,但它们的限制性位点不存在于 dbDNA 84中。反应产物的消化释放出 dbDNA 84和不需要的线性化质粒骨架85。随后用核酸外切酶消化该混合物,产生核苷酸、酶和缓冲液的混合物,可以很容易地将其与所需产物分离。dbDNA 84然后纯化以提供最小的线性 DNA 载体,编码几乎任何感兴趣的长序列。这些序列可以是复杂的或不稳定的,并且可以使用相同的过程重新扩增以快速生成数克数量的大 DNA,没有细菌或内毒素污染。作为制造平台,这种方法允许以比传统发酵方法快五倍的速度生产大 DNA(表 1)。 商业模式和去技能化 结合化学和酶促合成、序列选择和组装的方法将不断发展。然而,由于合成 DNA 的基础化学不太可能发生显着变化,延伸循环效率仍然是主要的限制因素。这促使公司开发高度并行化、小型化和自动化合成等互补能力,同时促进用户在生产 DNA 方面的自主权(图 7)。 图 7:DNA 合成中的用户自主权。
DNA 合成技术与开发这些方法的不同公司当前产品的复杂程度(左轴)以及这些公司提供的用户自主权(去技能化)(右轴)相对比,从专家参与到最终用户的完全自主权。例如,不依赖于模板的酶促寡核苷酸合成 (TiOES) 是 DNA Script 的基本技术,他们提供定制和多序列,同时将自动化集成到他们的合成工作流程中,从而开发出最终用户可以购买和使用的台式 DNA 打印机,只需最少的专家参与。Gibson,Gibson 组装方法;POS,亚磷酰胺寡核苷酸合成;RCA,滚环放大。 自动化与服务 研究重点是改进 DNA 序列的并行合成。众所周知,这会增加出错的可能性,特别是对于难以扩增的序列,例如重复序列或富含 GC 的序列。自动化提供了一个引人注目的方向。公司利用电子和微流体等其他领域的进步来改进 DNA 合成。与非自动化方法相比,这有助于识别和消除错误,提高准确性、规模和速度。例如,Evonetix 开发了一个用于高保真和快速基因合成的平台,该平台由硅芯片上数千个独立反应位点中的每一个以高度并行化的方式进行电化学处理来控制。并行合成和位点特异性热控制的结合有可能解决困难序列的局限性。例如,具有高 GC 含量的序列需要比其他序列更高的解链温度,并且可以形成稳定的二级结构,可以在高温下合成。然而,当使用这样的温度时,高位点特异性对于防止错误退火是必要的(表 1)。 现在有几家酶公司活跃在这个领域,出现了各种商业模式,越来越重视 DNA 组装和台式打印机。例如,作为台式 DNA 合成器提供的 DNA Script Syntax仪器的大小与 Illumina 开发的 HiSeq 测序仪的大小相似。该合成仪可在 6 小时内生成纯的 60 bp 寡核苷酸,可立即使用。 由于在提高延伸循环效率方面取得了适度的进展,因此正在开发使用微阵列技术来并行生成多个序列。尽管开发这些技术可能需要进一步投资,但它们将改进 DNA 合成。尽管可以预期新的、更有效的技术将推动和主导市场,但不太可能出现一种由一家供应商追求的成功技术。最终,几种类似有效技术的开发将确保所有供应商对 DNA 定价相似,例如,按基因或长度定价,从而使最终用户能够负担得起无限长度 DNA 的供应。每项技术都是专家开发的问题,但最终必须自动化,减少最终用户对专家参与的依赖,实现去技能化 DNA 合成(图 7)。 客户进入壁垒 定制 DNA 合成仍然是一项昂贵的工作(例如,每个 3 kb 基因 300-1,000 美元或 0.1-0.3 kb -1美元)。价格因供应商、序列组成和长度而异。观察到基因合成的价格在几年内下降到 0.01 kb -1美元的总体趋势,例如,对于基因片段,Twist Bioscience 提供的当前价格是 0.07 kb -1美元。需要更多的资金来帮助制造大 DNA 的研究。最终用户需要更专业的设备来制造比质粒更复杂的 DNA。可以将此类复杂和大型 DNA 的供应外包给 DNA 合成供应商(例如,用于组装的 Ribbon Biolabs)。为特定应用制作的定制 DNA 的复杂性决定了合成所需的技能障碍。一般趋势是通过减少对 DNA 合成进行故障排除的需要来减少对专家参与的依赖,这是通过酶性能和 DNA 组装方法的进步来实现的。 大量获取 DNA 的途径得到改善,基因组大小 DNA 的信息容量得到增强,可能会促进进一步的需求。因此,对可用长度的限制仍然是扩大规模的主要改进领域。一旦克服了序列的长度限制,预计对大 DNA 的需求将会增加。利用自动化芯片上基因组装的方法是有前途的解决方案。更长的 DNA 生产成本更高。然而,可以合理预期,随着更多技术能够突破长度限制,更多公司能够提供大的长的 DNA,合成 DNA 的价格将被压低。DNA 存储应用可能会提供例外情况,因为这些应用需要大量的起始材料(g kg −1) 产生比生物学应用所需的更大的 DNA。所需 DNA 的数量和长度与要存储的信息量有关。 实验室要求 越来越多的台式打印机将使内部 DNA 合成变得可行:Cytiva 的 ÄKTA Oligopilot 每 3-4 小时运行提供多达 8 个寡核苷酸,Kilobaser 每 2 小时运行提供 2 个寡核苷酸,而第一台酶促打印机 Syntax 可在6 小时内平行生产96寡核苷酸。通过服务提供商订购并在内部操作的 DNA 可以在小体积反应中组装,有助于通过小型化缩小实验规模。组装方法的自动化、小型化和并行化带来了降低 DNA 构建成本和缩短交货时间的更多好处,而组装 DNA 的准确序列验证则受益于 NGS 的功效。然而,入门级 DNA 合成实验室的保守估计起价为 20万美元,这可能会增加,具体取决于所需 DNA 的长度和生产规模。 在这方面,生物铸造厂为最终用户提供了互补的基础设施支持。这些设施建立在强大的高通量处理和分析技术平台之上,可以为长链双链 DNA 和多样化的变异库建立快速的内部生产管道。Biofoundries 通常托管 SynBio 堆栈的一个版本——一个技术生态系统,它允许通过将复杂任务分解为更小的任务来处理复杂任务,在自动化工作流程中提供这些任务的背景和目的以进行操作、组装、分析和组织小体积和高通量的 DNA 。 DNA 存储和可访问性 从第一本用 DNA 编写的书开始,人们一直对应用 DNA 来存储和保存社会不同部门产生的数据感兴趣。DNA 中永久、紧凑和低能量数据存储的概念受到关注,特别是通过 DNA 数据存储联盟——一个寻求使用 DNA 作为媒介创建数据存储生态系统的行业协会。 感兴趣的数据被编码在 DNA 的四个字母表中(即 A、C、G、T),而一组高保真酶用于创建这些数据的副本,并使用精确的测序技术检索它。例如,Catalog Technologies 和 Cambridge Consultants 构建了一个 DNA 合成器,它能够对 16 GB 的维基百科数据进行编码。随心所欲地为数据存储设计编码规则的能力可能会提供一种优雅的方式来适应错误率或避免特定的序列基序,这对于给定的合成或测序技术来说可能是困难的。例如,Hachimoji 碱基最近对遗传字母表的扩展创造了用八个而不是四个 DNA 字母表的字母编码数据的前景。这使得数据密度呈指数级增长,在工程酶的帮助下整合、复制和读取这种扩展遗传密码的碱基。这些发展意味着 DNA 数据存储产品成为可能,并且可能与生物应用相媲美,成为 DNA 合成技术的主要用途。 监督和标准化 制造长 DNA 片段的相对较新的能力可能会对基因操作和对生命系统的控制产生影响,这需要在全球范围内进行监督和监管。相关领域制定了监管政策;例如,通过关于重组 DNA 研究的 Asilomar 会议,社区在会上引入了生物安全法规的自我监管程序。最终,非专家也能获得编写 DNA 的能力。因此,人们越来越认识到需要制定监督政策来减轻滥用 DNA 技术带来的生物安全风险。新政策的引入有助于调整现有机制以评估这些和不断变化的风险. 例如,一个国际基因合成联盟由行业组成,旨在开发一个通用协议来筛选合成序列,以及订购这些序列的客户,从而自我调节序列身份。然而,DNA 技术的进步速度超过了理解、监控和调节风险的能力。这类似于 CRISPR 基因编辑,其中在监管机构了解滥用该技术进行人类基因组编辑的后果之前,最终用户已经具备了基因组工程能力。 当天然 DNA 被纳入生物体后可以自我维持,生物体随后可以永久地嵌入生态系统,从而引发围绕水平基因转移的问题作为进化的关键部分. 因此,需要对与 DNA 合成相关的道德因素、法规和其他风险进行深思熟虑的回应和监督。同样,最大限度地提高 DNA 合成的可重复性和可靠性也至关重要。尤其重要的是要满足新兴法规的要求,这些法规要求行业证明其产品和技术的可追溯性。这种对可重复性和可追溯性的日益关注优先考虑了标准化的需要。缺乏可用于评估合成 DNA 和合成方法(包括 XNA 等新型化学物质)的性能和质量的参考材料和方法是一项关键挑战。参考材料可以包括 DNA 序列、个体或文库,它们可追溯到国际单位制 (Système International d'Unités — SI)。参考方法可以提供合成程序来对商业方法的性能进行基准测试,例如,与延伸循环效率有关。令人鼓舞的是,参考材料已经面世,例如美国国家标准技术研究院的第一个“人类基因组”DNA 参考材料(RM 8398),它可以评估 NGS 检测的准确性。需要新的计量学为 DNA 合成提供比较和再现性的基础,以支持现有和新兴的 DNA 合成技术。 结论 随着能够读取 DNA 的技术的快速发展,写入 DNA 的能力已经落后。迄今为止开发的 DNA 合成技术在弥合 DNA 写入差距的能力方面可能有所不同(表 1和图 1)。然而,它们的持续发展是由两个主要因素驱动的:缺乏常规地以规模和成本制造无限长度 DNA 的方法,以及不同和不相关部门对 DNA 的需求不断增加。随着 NGS 的成功,这两个因素刺激了对创新技术的探索,并保证了任何能够克服大小限制 DNA 合成障碍的策略的商业化成功。因此,DNA 制造商严密保护他们的知识,并对他们的技术可以提供什么的声明持谨慎态度。这是值得注意的,因为大多数现有方法都是相似的,使用相同的起始材料,这表明创新进展缓慢。相反,激烈的竞争促使公司在早期阶段寻找应用领域或展示其技术在生产具有挑战性的 DNA 分子中的用途。 自动化的进步将使 DNA 合成越来越容易为非专家所用。大多数供应商,尤其是那些提供合成 DNA 作为服务的供应商,都意识到需要监督和监管政策来保护他们的商业和声誉利益,并可能反过来为此类政策的制定做出贡献。一旦 DNA 合成对于制造新 DNA 分子的爱好者的小型黑客空间来说是负担得起的,所产生的 DNA 的用途将难以控制。旨在根据适用政策和道德规范监控合成 DNA 的使用和分发的风险治理将减少不良事件的可能性。 总而言之,新的合成方法将继续出现,并持续关注提供更环保的解决方案,减轻因使用有机溶剂和危险化学品而对环境造成的潜在有害后果。由于现有合成方法的局限性,不太可能很快出现有效合成无限大小 DNA 的常规方法。然而,在可预见的未来,基因写入缺口仍有很大的突破空间。 |

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号