来源:小桔灯网 作者:奔跑的骆驼 “ 前言:2021年10月1日,新版的《体外诊断试剂注册与备案管理办法》即将实施。新旧版本些许措辞的调整背后,蕴含着诸多管理理念的优化和执行层面的细化。 ☞ 举个例子:开篇宗旨旧版讲保证IVD的安全、有效(全文共提到安全3次、有效4次),新版讲保证IVD的安全、有效和质量可控(全文提到安全29次、有效40次、质量可控10次),在安全和有效的基础上将质量可控这个维度单独强调出来,管理理念越发成熟全面。 我们总强调IVD的安全性和有效性,那么安全有效的内涵和准则究竟是什么?本文小编针对第14条的要求做简要梳理和延伸,希望能够带给各位桔友些许启发,限于阅历和能力,不正之处恳请批评指正。
➣ 第十四条的变化 先来看原文: 第十四条 体外诊断试剂注册、备案,应当遵守相关法律、法规、规章、强制性标准,遵循体外诊断试剂安全和性能基本原则,参照相关技术指导原则,证明注册、备案的体外诊断试剂安全、有效、质量可控,保证信息真实、准确、完整和可追溯。 (旧版:第十三条 申请人或者备案人申请注册或者办理备案,应当遵循体外诊断试剂安全有效的各项要求,保证研制过程规范,所有数据真实、完整和可溯源。) 与旧版第十三条相比,新版要求更加细致明确:我们需要通过遵守法律法规、遵循IVD安全和性能基本原则、参照技术指导原则这一系列的动作,来证明IVD的安全、有效、质量可控。 IVD安全和性能基本原则就此被明确提出,它相当于IVD技术文件中的宪法,堪称安全有效的万源之本,企业产品研发、生产,监管部门评审,都应依据它。既然基本原则这么豪横,我们就好好的扒一扒。 ➣ IVD安全和性能基本原则的由来 1992年,美国、欧共体、澳大利亚、日本和加拿大共同发起了一个非官方性国际组织-全球医疗器械协调工作组(GHTF)。GHTF的目标是在医疗器械管理体系的发展过程中,鼓励全球管理水平的趋同,以最合适的管理方式处理保障公众健康问题。实现目标的方式主要是制定一系列指导性文件。 2005年,GHTF首次发布了《Essential principles of safety and performance of Medical devices and medical devices and IVD medical devices》(简称EP),并与2012年进行了更新。 2011年,来自美国、加拿大、欧盟、日本、澳大利亚和世界卫生组织(WHO)医疗器械监管机构成立国际医疗器械监管机构论坛(International Medical Device Regulators Forum,IMDRF),在GHTF的工作基础上,由来自全球的医疗器械监管者自愿成立的组织,旨在推动全球医疗器械安全标准协调统一,从而加速国际医疗器械监管的协调、融合。2013年国家食品药品监督管理总局成为IMDRF正式成员。
2018年IMDRF对《EP》进行了再次修订和完善。(2018年由中国担任IMDRF轮值主席国,所以我们看到2018版的EP发布签名是轮值主席国主席、国家药监局袁林司长。)
医疗器械产品种类繁多,产品的安全有效性千差万别。医疗器械注册并非是对标准符合性的审查,标准仅是技术审评过程中的要素,是对单一或者部分风险控制的方法或者部分有效性的评价方法,完成标准的评价只是系统评价中的一部分。没有一个标准、指南能够涵盖不同产品的评价要求。对医疗器械产品的评价,必须结合产品的特点进行全面的安全有效性评价。 为了达到这一目标,国家药监局充分吸取借鉴国际医疗器械监管经验,对2018版《EP》等同转化,在2020年3月发布了中文版《医疗器械安全和性能基本原则》。 医疗器械产品的审评审批工作正是基于《医疗器械安全和性能基本原则》,根据申请人提交的注册申报资料,针对具体产品特点,基于科学原则,对产品的安全性、有效性、质量可控性开展的系统评价。 ➣ IVD安全和性能基本原则的基本内容
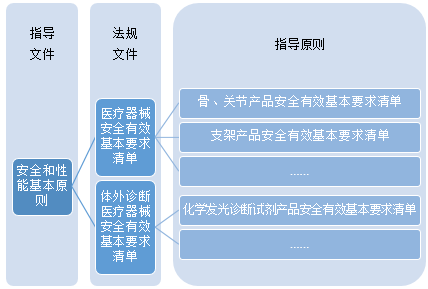
《基本原则》共78个条款,可分为三个层级:
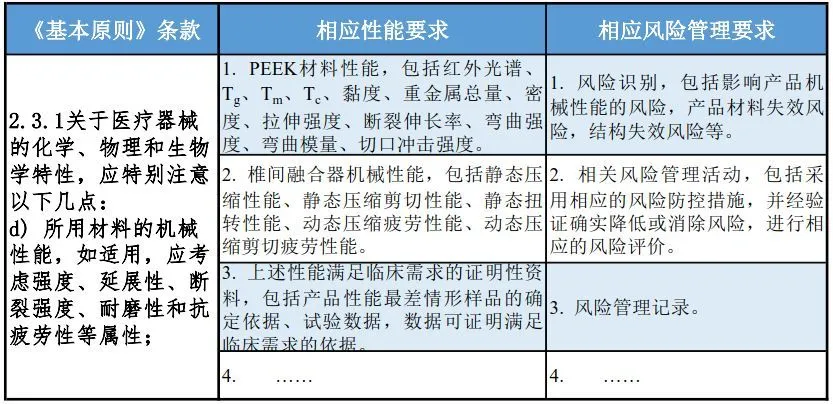
➣ 基本原则的应用举例 医疗器械千差万别,需结合不同的产品特点,根据《基本原则》,明确产品在设计、生产等环节中应满足的性能,识别相关的风险,采取相应的控制措施降低风险。IVD的成熟案例小编手头还没有,先分享两个医疗器械的侧面学习一下: ◇ 第一个是在机械性能和风险角度的应用(PEEK脊椎融合器)
◇ 第二个是在质量体系构建方面(无菌金属骨科植入物)
前面讲到,基本原则不止针对注册人/备案人,也同时适用于注册审评。基于《基本原则》将会延伸出具体的医疗器械和IVD安全很性能基本要求清单作为第二级法规文件;而针对不同的产品又会细化所应建立的体系文件、风险管理以及性能评价,形成第三层级指导原则。
➣ IVD安全和性能基本原则清单的获取 今年药监局在《体外诊断试剂注册申报资料要求和批准证明文件格式(修订草案征求意见稿)》中(下图二维码链接),附录6部分正是《体外诊断试剂安全和性能基本原则清单》,详细随着注册管理办法的实施,配套资料要求将会很快发布,敬请关注。
结语 安全(定义为“无不可接受风险”)有效(定义为“医疗器械在绝大多数目标人群中具有显著临床效果的能力”)的医疗器械是通过包括产品设计开发在内的完善的质量体系和贯穿始终的风险管理实现的,在整个风险管理过程中,需要针对产品的预期用途和使用条件积累充足的风险及受益相关的科学证据,这是医疗器械产品安全有效的内部保障。 安全有效是医疗器械技术审评的核心。《医疗器械安全和性能基本原则》,规定了为实现在全生命周期内均能达到预期安全和性能要求的医疗器械产品,注册人/备案人所需达到的基本的设计和生产要求。 需要强调的是,该原则内容涉及医疗器械产品安全和性能的基本要求;针对不同的产品,还有更多具体和全面的要求需要满足。对于产品安全有效的判定最终仍是要基于风险/受益比,即风险可接受原则。 随着新的注册管理办法的实施,详细很快就会有更多关于《IVD安全和性能基本原则清单》更加专业和深入的解读发布出来,小编抛砖引玉,跟大家简单分享,不正之处还请批评指正。 |


 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号