登陆有奖并可浏览互动!
您需要 登录 才可以下载或查看,没有账号?立即注册


×

当找到有效的活性分子后......
这些分子的作用靶点是什么?
如何发挥它们的生物学效应?

本期咱们一起探讨药物找靶的新策略,Let’s Go!
Section.01 药物靶点的识别
明确药物的临床适用范围是药物发现与设计的关键,除此之外,阐明药物的作用靶点也在疾病的发生发展中扮演重要角色。
诺贝尔生理学或医学奖得主 Paul Ehrlich 曾说过“corpora non agunt nisi fixate”—— 药物不会发挥作用,除非它们被结合。《牛津生物化学与分子生物学词典》将药物靶点定义为与特定化合物相互作用并受其活性调节的生物实体,通常是蛋白质或基因。
01有潜力的药物靶点都有哪些特点[1]?1. 在某种疾病的病理生理中有确凿的作用,和/或能够改变疾病进程; 2. 在全身范围内的非均匀分布; 3. 有可用于评估成药性的三维结构; 4. 易于进行“检测”,便于高通量筛选; 5. 具有良好的毒性特征,可以通过表型数据预测潜在的不良反应; 6. 具有有利的知识产权 (IP) 状态 (这一点主要与新药研发公司相关)。
新药发现主要分为基于靶点的药物发现 (Target-based drug discovery, TDD) 和基于表型的药物发现 (Phenotypic drug discovery, PDD)。对于 PDD 来说,从大规模化合物中筛选并验证得到活性分子后,进一步发现并阐明其作用靶点更是至关重要!
药物靶点识别的方法主要分为计算机预测与实验找靶。
计算机预测通常利用数据挖掘、药效团模型匹配、正反分子对接技术和药物-靶标相似性算法等,识别小分子可能作用的靶点。下面是一些常用的靶标预测网站,有需可参考喔~
表 1. 常用的靶标预测网站。    
虽然通过预测的方法能够快速筛选出大量潜在靶点,但精准度相对不高,而且通常无法定位到某种细胞或组织中。由于靶点可能在不同的细胞中表达水平不同或发挥不同的作用,这种缺乏细胞特异性的预测可能会导至在实验验证阶段的失败。
实验找靶则是通过具体的生物实验来筛选靶点,其底层逻辑一般是通过各种实验方法找到细胞或组织中能够与药物结合的潜在蛋白,而后通过结合层面和功能层面的双重验证来确认靶点在疾病中的作用以及药物可以通过调节该靶点功能从而发挥药效。这不仅能够提高靶点筛选的准确性,也为靶点选择提供了更丰富的生物学背景,使得药物研发更具针对性和有效性。
实验找靶常用的方法主要有 DARTS、SPR 和 Pulldown 技术等,且看小 M 为大家逐一介绍~
Section.02 DARTS 找靶技术
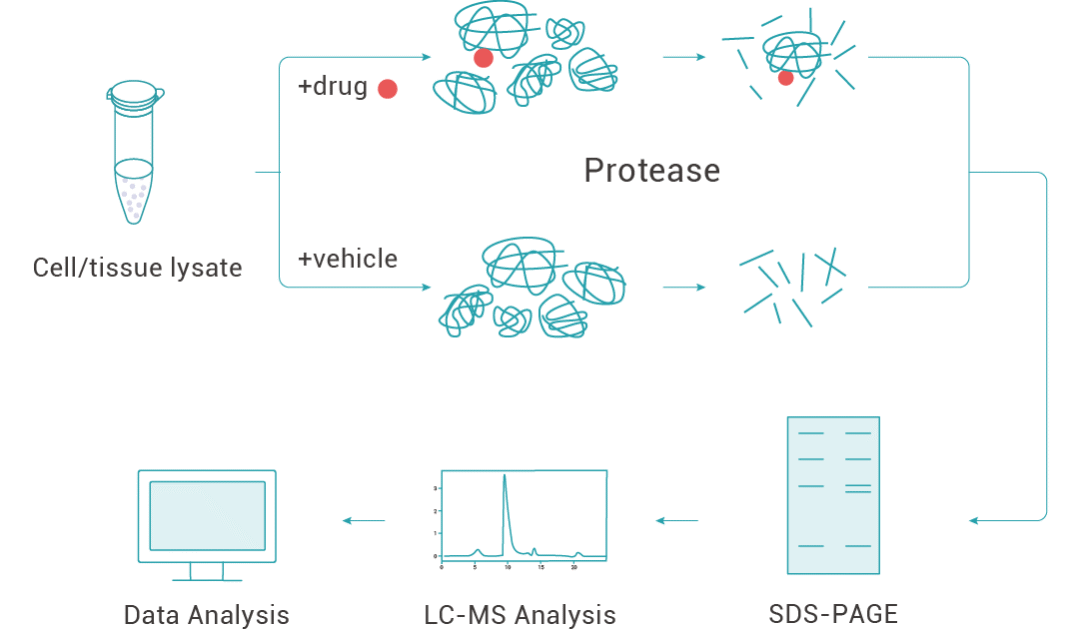
药物亲和反应靶点稳定性(Drug affinity responsive target stability, DARTS)的概念最初由 Brett Lomenick 等人于 2009 年提出,其基本原理是靶蛋白与小分子配体结合后稳定性增加,可以增强靶蛋白对蛋白酶酶解作用的抗性,以此来进行靶蛋白筛选,同时无需对小分子进行修饰[2]。
DARTS 找靶的一般实验流程:
1. 蛋白库的制备 (通常为目标细胞或组织的蛋白裂解液); 2. 蛋白裂解液与小分子共孵育; 3. 蛋白酶水解 (合适的蛋白酶浓度至关重要); 4. 通过考马斯亮蓝染色或银染等方法检测对照组和加药组蛋白的差异; 5. 目标蛋白凝胶条带收集; 6. 通过质谱等方法对目标蛋白进行鉴定。
图 1. 利用 DARTS 技术找靶的流程。
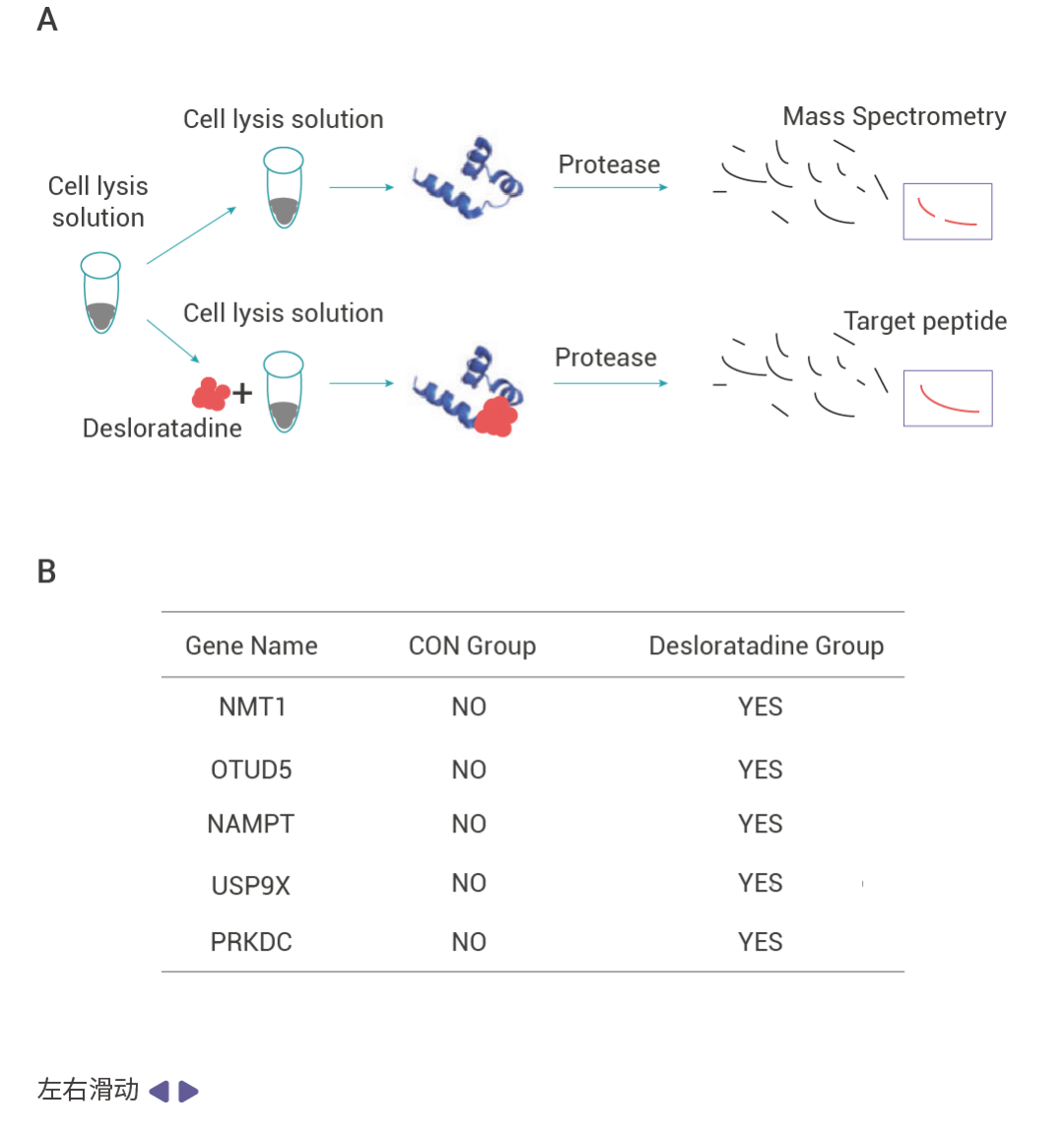
文献案例作者通过表型筛选及验证发现药物 desloratadine 在体内和体外实验中都能够显著抑制肝细胞癌 (HCC) 的生长和增殖。为了探究 desloratadine 发挥抗癌生物活性的分子机制,作者通过 DARTS 实验发现了 53 种潜在靶蛋白,并从中选择了 5 种感兴趣的蛋白质修饰酶进行进一步研究。通过蛋白表达、Knockdown、细胞增殖、细胞侵袭和迁移以及体内实验等验证了 desloratadine 通过靶向 NMT1 蛋白调节 HCC 的发展,利用 SPR 技术验证了 desloratadine 和 NMT1 蛋白的结合,通过分子对接、蛋白点突变联合 SPR 技术找到了 desloratadine 和 NMT1 蛋白的结合位点。 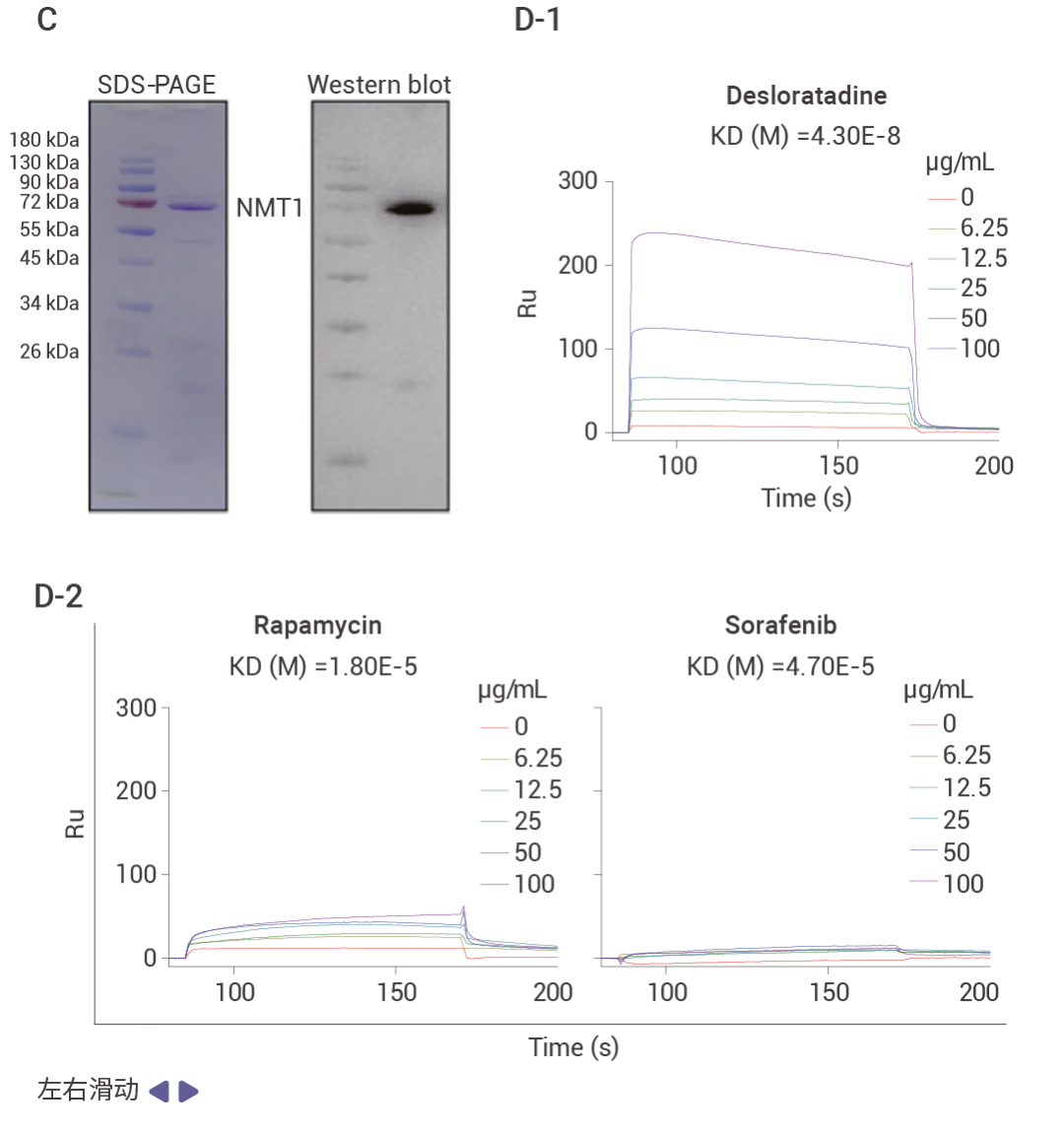
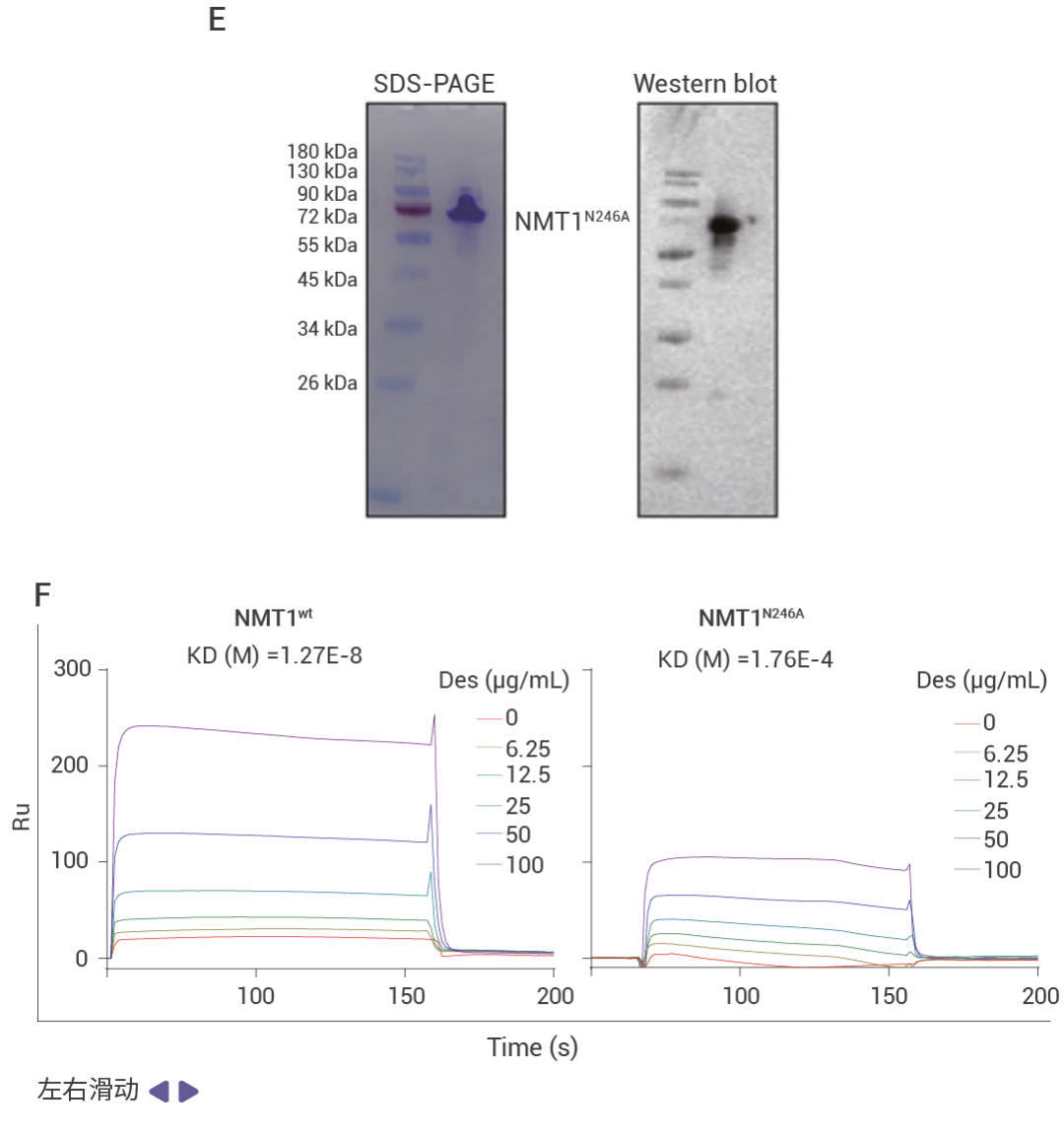
图 2. 利用 DARTS 技术发现 desloratadine 通过靶向 NMT1 蛋白调节 HCC 的发展[3]。 Section.03 SPR 找靶技术
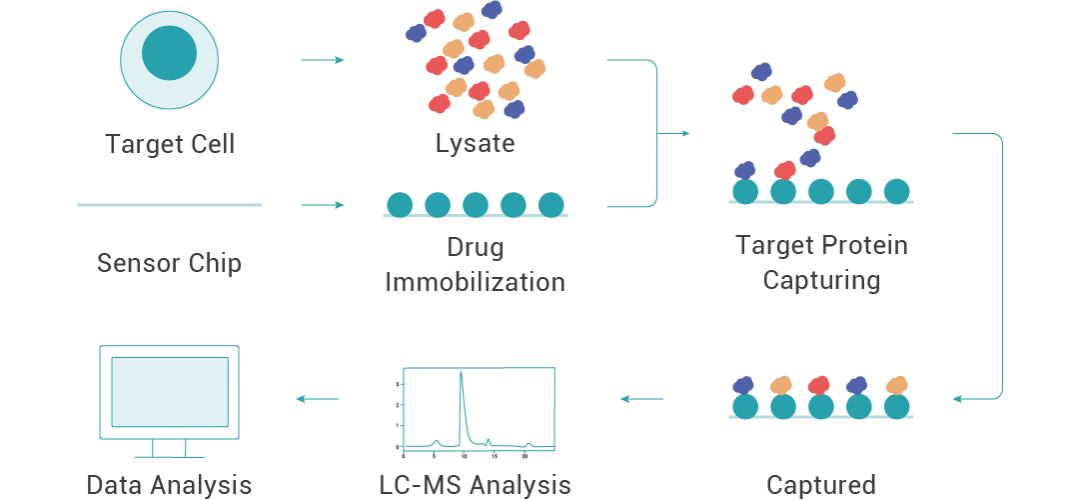
表面等离子共振 (Surface Plasmon Resonance, SPR) 是一种无标记、实时检测分子相互作用的技术。它基于表面等离子共振原理,实时监测分子在固体表面 (通常是金属表面) 上的结合与解离过程。
虽然 SPR 实验通常用来检测两个分子之间的相互作用,但是 SPR 的这一特性也使其可以用于从复杂样品中“垂钓”与药物相互作用的分子。
实验时,将药物固定在芯片上,蛋白裂解液稀释为不同浓度,采用多浓度上样的方式,将不同浓度的裂解液与药物互作,收集与小分子结合的蛋白并通过高分辨质谱进行鉴定,从而得到药物的潜在靶蛋白。同时,SPR 技术也常用于靶蛋白与药物的亲和力验证。
图 3. 利用 SPR 技术找靶的流程。
文献案例作者通过细胞活力、细胞凋亡等表型实验从 1,504 种 FDA 上市药物中筛选并验证了 Lomitapide 对耐药多发性骨髓瘤 (Multiple myeloma,MM) 细胞有很好的抗肿瘤活性,体内实验也证实 lomitapide 可以抑制 MM 的疾病进程。
为了确定 lomitapide 的潜在靶点,作者利用 SPR 结合 HPLC-MS 筛选得到 39 个可以与 lomitapide 结合的蛋白,并选择了 MS 分析排名第 1 位的 PARP14 蛋白进行验证。通过 SPR 实验证实 lomitapide 与 PARP14 蛋白能够直接结合 (Kd = 101 nM),并在后续实验中利用 knockout 证实 Lomitapide 通过靶向 PARP14 诱导 MM 线粒体功能障碍和线粒体自噬,从而揭示了 Lomitapide 对抗 MM 的新机制。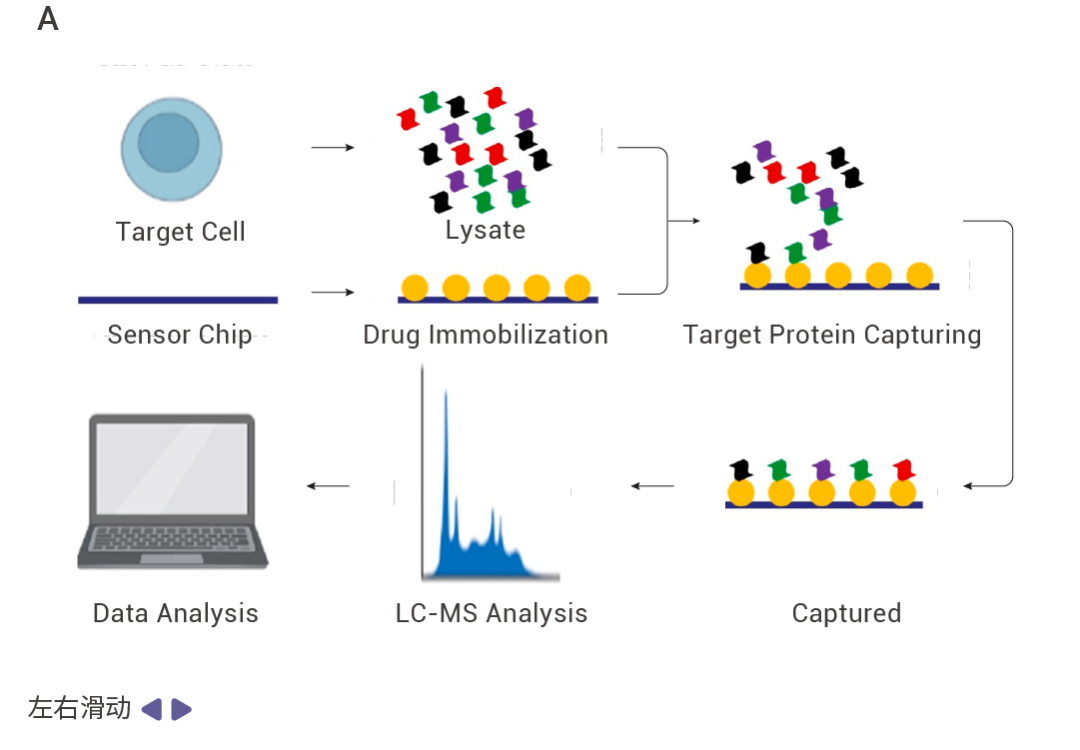 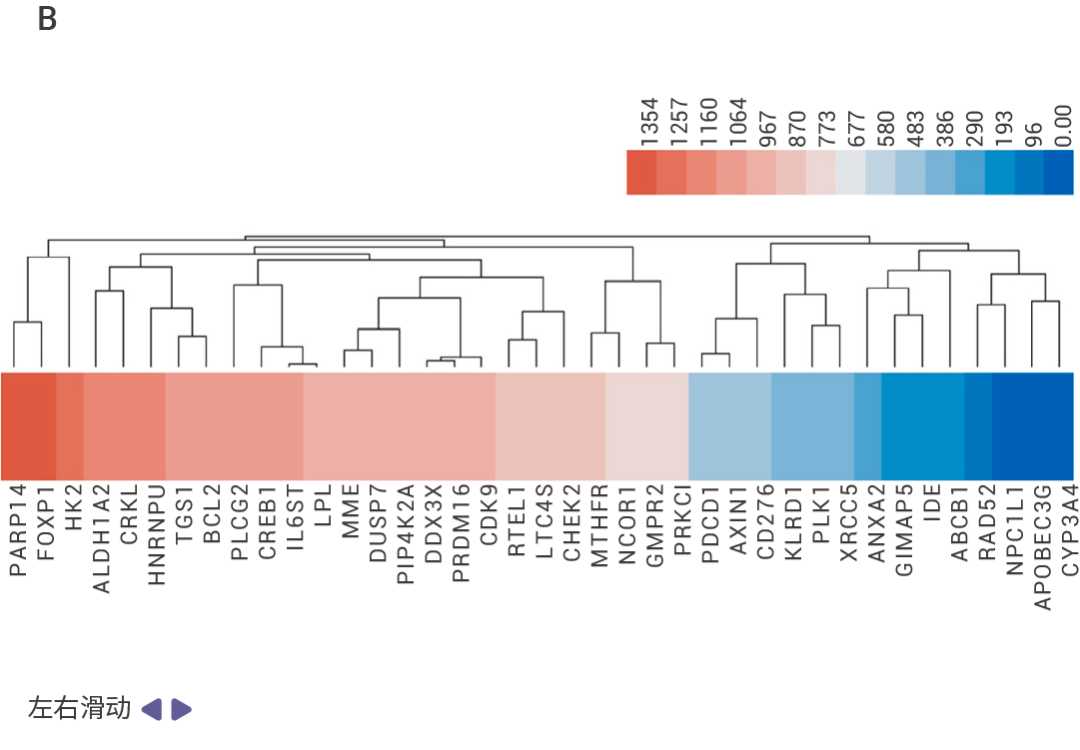 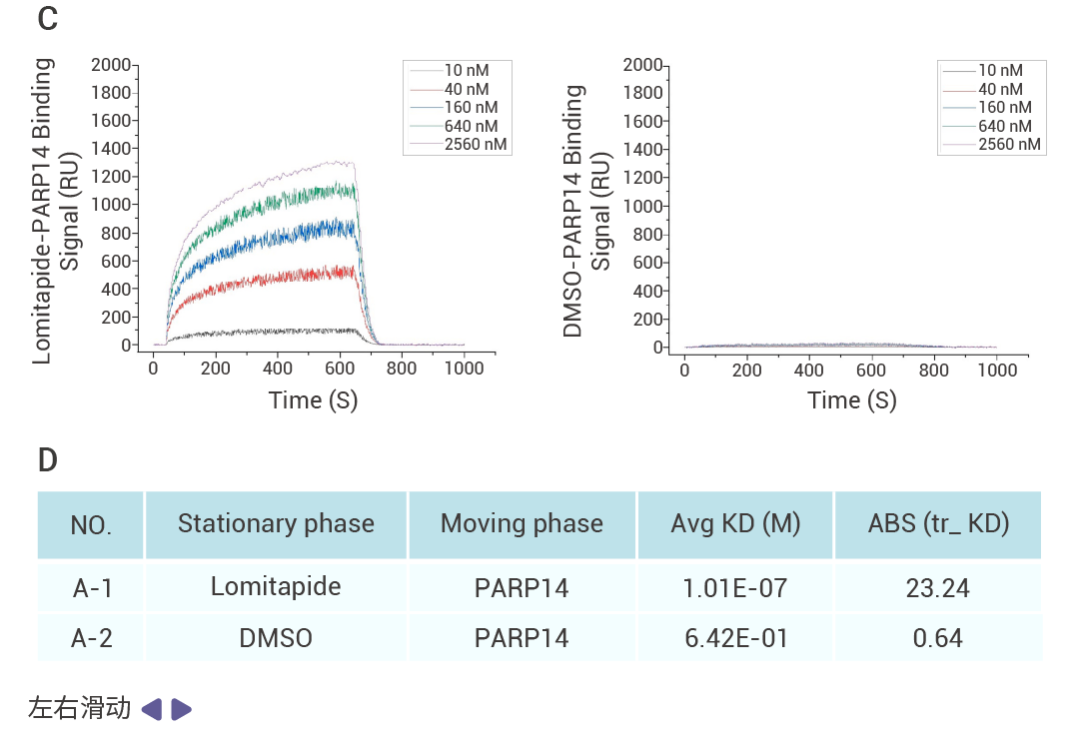 Section.04 SPIDER 邻近标记 Pull Down 技术
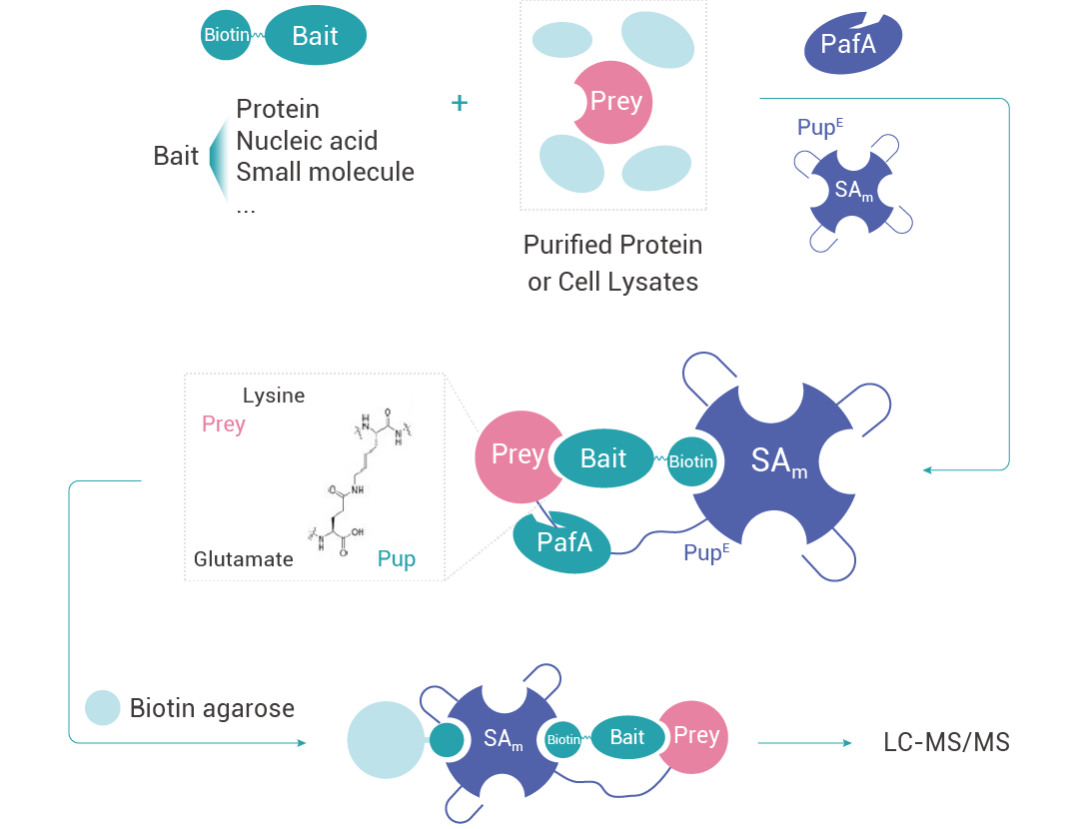
特异性糖基化作为标识报告物 (Specific Pupylation as IDEntity Reporter, SPIDER) 是一种基于底物的邻近标记活性和链霉亲和素 (SA)-生物素系统来识别蛋白质-生物分子相互作用的方法。
当靶蛋白 (Prey) 与带有生物素标记的诱饵分子 (Bait) 发生相互作用时,会同时与生物素偶联上的 SAm-PupE 靠近,在 PafA 酶的催化作⽤下,PupE 的 C 末端会与 Prey 蛋白表面的赖氨酸残基共价相连,从而实现对靶蛋白的捕获。该过程可以将生物分子与蛋白质之间的非共价结合相互作用转换为 SAm-PupE 与靶蛋白的共价连接,从而能够稳定地用于后续靶蛋白的富集、鉴定和分析。
图 5. SPIDER 技术原理示意图。
对于实验找靶而言,针对药物特点选择合适的实验方案尤为重要,一起来看看上述 3 种药物找靶技术各自的应用范围吧!
表 2. 不同药物找靶技术的特点及应用范围。  
那么有的小伙伴可能会问了,找到潜在结合蛋白之后,要如何验证这些靶点是否是疾病的靶点呢?活性分子又是否是通过某个靶点发挥药效的呢?别着急,后续的验证方法小 M 也为你总结好了,噔噔噔噔~
表 3. 实验找靶后续验证方法举例。
看了小 M 的介绍,大家现在有没有一些找靶的思路了呢?MCE 药物找靶平台提供多种找靶技术,结合经验丰富的蛋白表达纯化和分子互作研究平台,可以为药物潜在靶点的识别和验证提供全面和个性化的实验方案。欢迎来和小 M 一起探讨~
[1] Isabella Gashaw, et al. What makes a good drug target?. Drug Discovery Today. 2012.S24-S30. [2] Lomenick B, et al. Target identification using drug affinity responsive target stability (DARTS). Proc Natl Acad Sci U S A. 2009;106(51):21984-21989. [3] Tan, XP., et al. Blockade of NMT1 enzymatic activity inhibits N-myristoylation of VILIP3 protein and suppresses liver cancer progression. Sig Transduct Target Ther 8, 14 (2023). [4] Honghao Zhang, et al.Targeting PARP14 with lomitapide suppresses drug resistance through the activation of DRP1-induced mitophagy in multiple myeloma. Cancer Letters.2024.
|

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 






 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 发表于 2025-5-29 14:44
发表于 2025-5-29 14:44