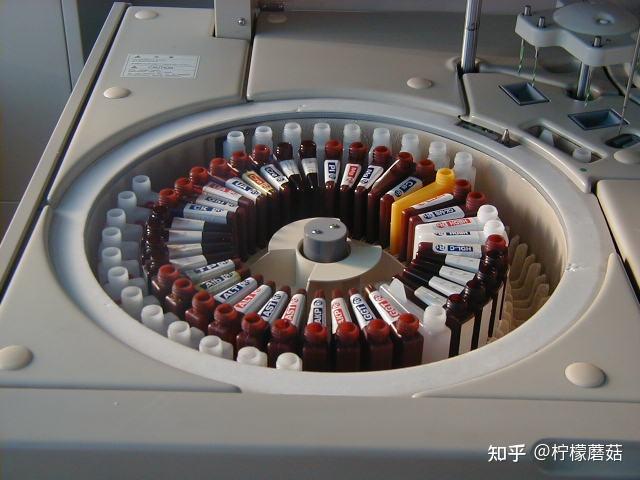
样品系统
样品包括:校准品、质控品、临床标本。
由样品装载、输送和分配装置组成。
b. 试剂系统
由试剂储放和分配加液装置组成
c. 反应系统
由反应盘、混合装置、温控装置组成
d. 清洗系统
由吸液针、吐液针和擦拭刷组成
e. 管路系统
包括向反应体系注入纯水或洗液的管路、连接注射器 与探针的管路、排除废液的管路。
f. 比色系统
光源:一般采用卤素灯
比色杯:硬质玻璃、石英和优质塑料
单色器与检测器
g. 程序控制系统 试剂仓
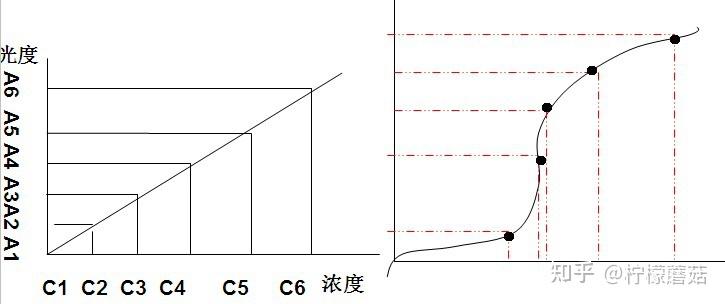
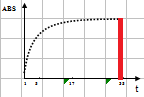
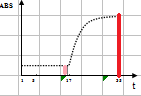
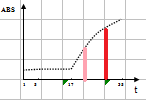
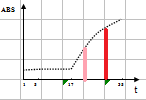
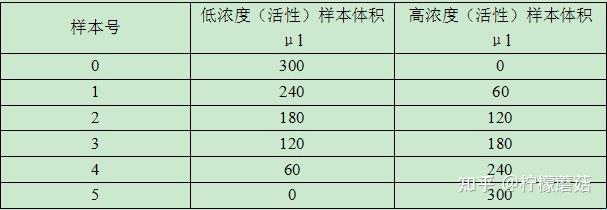
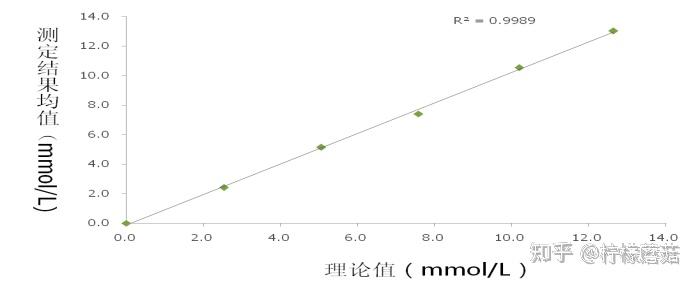
线性范围(linearity range ):在待测物质的一定浓度范围内,反应的吸光度值与待测物浓度成线性关系。试剂盒一般提供了线性范围参考值,但主张在本室条件下制作校准曲线,实测线性范围。测定浓度值超出设定范围即判定非线性。不光要注意线性范围上限,也要注意下限的设定。设定时应结合方法学(如校准曲线是否有截距,灵敏度和最低检出限) 、参考范围 (如 ALT 不可能为零值或负值,有临床意义的上下限值)等。

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 






 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 发表于 2025-5-11 16:00
发表于 2025-5-11 16:00