金桔
金币
威望
贡献
回帖0
精华
在线时间 小时
|
新药研发不仅看监管机构的水平,也需要有研发实力的医药人共同努力。
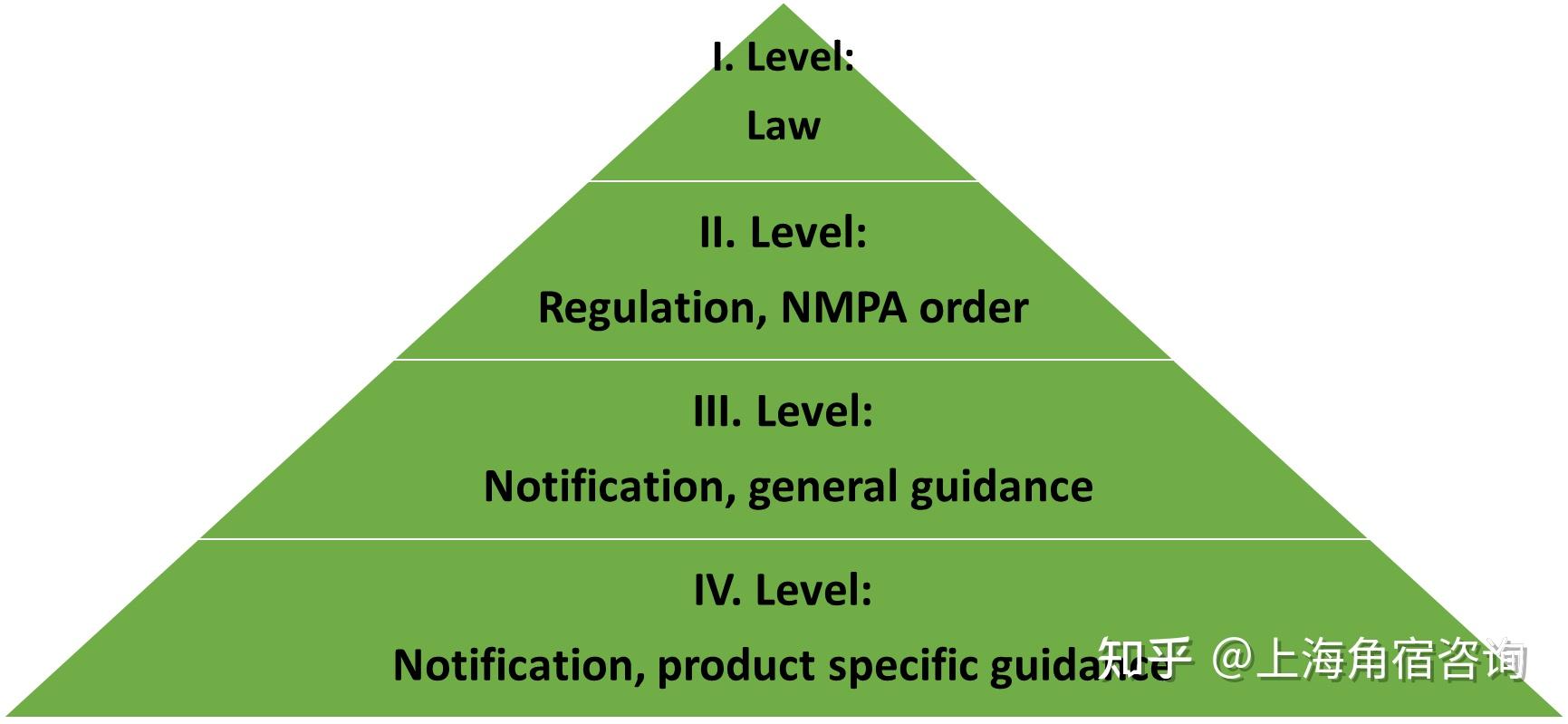
现阶段NMPA(CFDA)面对FDA,确实还是小宝。美国的药物研发还是先进很多,不论是first-in-class 的数量和品质,整体药物研发的数量和品质,先进评价方法的接受程度,都是全球领先的。当下我们NMPA制定的不少指导原则也是主要参考FDA相对应的指导原则草拟的,中国首批的全球新药的数量还是非常有限的。当然我们NMPA对于中药研发的监管和推动还是很有中国特色的,这部分是FDA所没有的。
我国的新药研发起步比较晚,不论NMPA、药企还是相关各方都还有较大的成长空间。我国大多的药企都是等美国的同机制的药上市之后才开始跟进,因此根据临床研发的周期,大约晚/慢10年。当然,我国医药人第一方队的成长速度也是喜人的,第一方队的新药研发能力基本是和国际第一方队持平的,但是第一方队的数量非常有限。
高新技术是国力的体现,我国自主研发的新药是人民健康的保障。随着国力的增强,NMPA的实力也会越来越强。我们没必要因为当下的境况就妄自菲薄,中国人最擅长的就是借他人的经验还帮助自己迅速提升。一起加油吧! |
|

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 






 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 发表于 2025-3-30 16:10
发表于 2025-3-30 16:10