金桔
金币
威望
贡献
回帖0
精华
在线时间 小时
|
基因测序是指对DNA进行高通量测序,以获得个体基因组的全面信息。通过快速、高效地分析大量的DNA序列,基因测序技术已经成为研究和诊断领域中不可或缺的工具。但是,基因测序产生的数据非常庞大,需要进行复杂的数据解读和分析才能得到有用的信息。
一、基因测序数据类型
首先,需要了解基因测序数据的类型。基因测序可以分为三种类型:全基因组测序(Whole Genome Sequencing,WGS)、外显子组测序(Whole Exome Sequencing,WES)和靶向测序(Targeted Sequencing)。其中,WGS测序对整个基因组进行测序,包括基因区域和非编码区域;WES测序仅测序编码蛋白质所需的外显子区域;而靶向测序则是针对特定基因或区域进行测序。由于不同类型的基因测序数据在数据量、覆盖度和数据结构等方面存在差异,因此需要根据实际应用场景选择合适的测序方法。
二、基因测序数据质量控制
在进行数据分析之前,需要对基因测序数据进行质量控制。基因测序数据的质量受多种因素影响,如DNA提取、文库构建、测序仪器和测序数据处理等。因此,在进行数据分析之前,需要对原始数据进行质量控制,以排除低质量数据对后续分析的影响。
常用的基因测序数据质量控制方法包括以下几个方面:
- 原始数据质量评估:利用FastQC或FASTP等软件对原始数据进行检查,可以评估数据的质量和数据结构等信息,帮助筛选出低质量的数据。
- 过滤低质量序列:通过Trimmomatic、BBDuk等软件对原始数据进行去除接头序列、过滤低质量序列和修剪末端碱基等操作,从而提高数据质量。
- 消除污染序列:使用Kraken或KneadData等工具对测序数据进行污染序列的消除,可以排除来自外源菌群、病毒或其他生物的序列,避免对后续分析造成干扰。
三、基因测序数据比对和变异检测
经过质量控制后,需要将测序数据比对到参考基因组上,以确定样本的基因型和变异信息。基因测序数据比对是指将测序数据与已知的参考基因组进行比对,从而确定样本的DNA序列在参考基因组上的位置、覆盖度和相似性等信息。
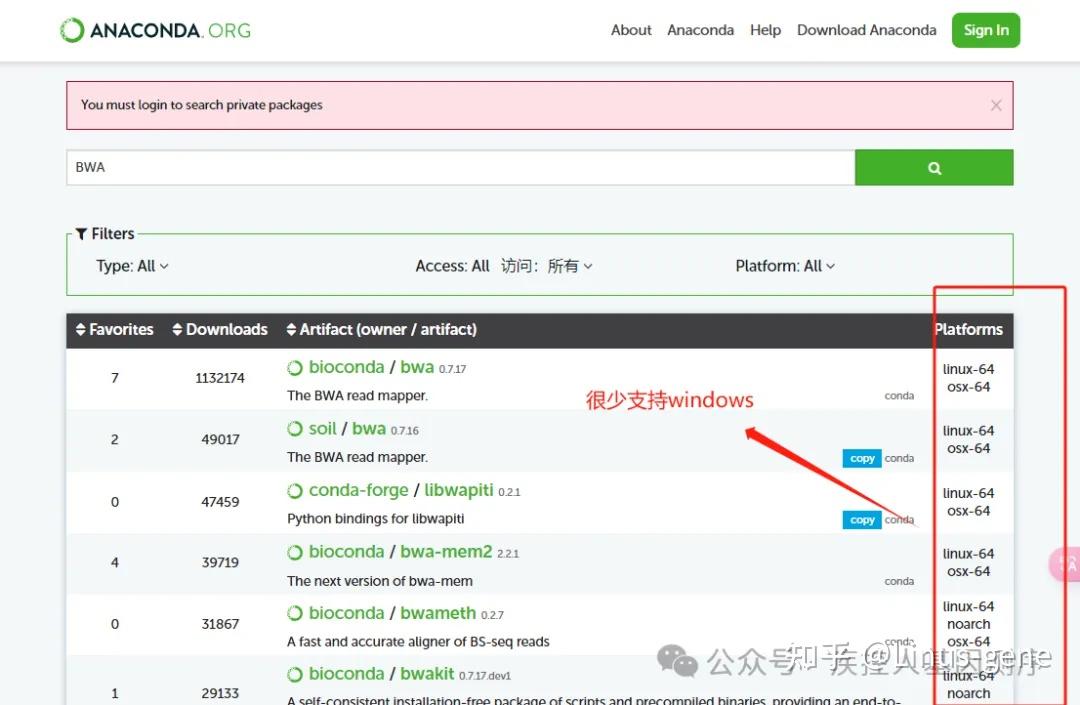
常用的基因测序数据比对软件包括Bowtie2、BWA、HISAT2等。这些软件采用不同的算法和策略,可以根据实际情况选择合适的软件进行比对。比对结果可用于检测SNP、Indel、CNV等遗传变异信息,为后续的生物信息学分析提供重要的依据。
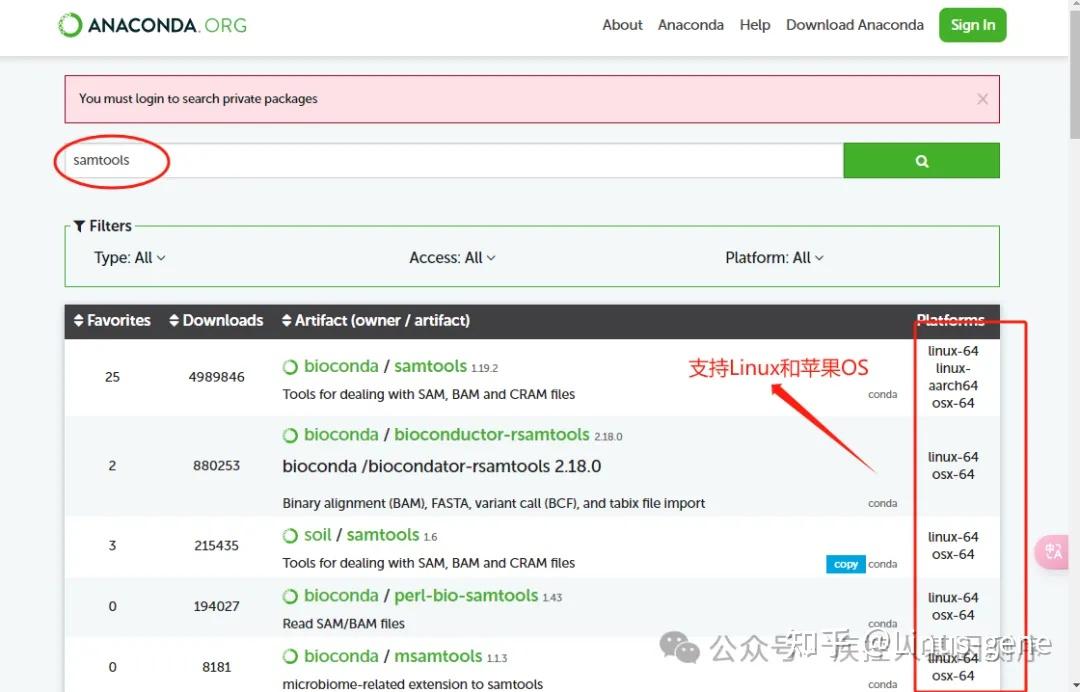
在进行变异检测之前,需要先对比对结果进行去重和排序,以便更好地进行后续的变异检测和注释。常用的去重和排序软件包括Picard、SAMtools、GATK等。
接下来,需要对比对结果进行变异检测,以确定样本与参考基因组之间的差异。变异检测可以分为单样本检测和多样本比较两种方式。
单样本变异检测是指将一个样本的测序数据与参考基因组进行比对,然后从比对结果中检测出该样本的变异信息。常用的单样本变异检测软件包括GATK、VarScan、FreeBayes等。这些工具可以检测出SNP、Indel、CNV等各种类型的遗传变异信息,并提供丰富的过滤方法和统计结果,帮助筛选出高可信度的变异位点。
多样本比较是指将多个样本的测序数据与参考基因组进行比对,并比较它们之间的差异,以确定不同样本之间的共有和私有变异信息。多样本比较可以帮助研究人员了解不同样本之间的遗传差异,并找到与某一特定表型相关的遗传变异信息。常用的多样本比较软件包括GATK、VarScan2、Strelka等。
四、基因测序数据注释和功能分析
在进行变异检测后,需要对变异位点进行注释和功能分析,以确定它们的生物学含义。基因测序数据注释是指将变异位点与已知的基因组注释信息进行比对,从而确定该位点的位置、影响和可能的功能等信息。常用的基因测序数据注释工具包括ANNOVAR、VEP、SnpEff等。
除了注释之外,还需要对变异位点进行功能分析,以了解其可能的生物学功能。常用的基因测序数据功能分析方法包括以下几个方面:
- 功能富集分析:通过对变异位点所在的基因进行富集分析,可以了解这些基因是否参与某些特定的生物过程或通路。
- 蛋白质结构预测:通过模拟蛋白质结构和功能,可以预测某些变异位点是否对蛋白质结构和功能产生影响。
- 代谢通路分析:通过对变异位点所在的基因进行代谢通路分析,可以了解这些基因是否参与某些重要的代谢过程。
- 突变的遗传学效应分析:通过对变异位点所在的基因进行遗传学效应分析,可以了解这些基因对特定表型的影响和可能的疾病相关性。
五、基因测序数据可视化和解释
最后,需要对基因测序数据进行可视化和解释,以便更好地理解和交流数据结果。常用的基因测序数据可视化和解释工具包括IGV、UCSC Genome Browser、Ensembl等。这些工具可以将测序数据和注释信息可视化为图形化界面,方便用户查看和分析结果。
除了可视化之外,还需要对基因测序数据进行解释,以便更好地理解结果和决策。解释需要结合实际情况和领域知识,对变异位点的生物学含义进行推断和判断。在解释过程中,需要考虑以下几个方面:
- 变异位点的频率和遗传学效应:根据变异位点的频率和遗传学效应,可以判断该变异是否与某个特定表型相关。
- 位点功能和位置:通过注释信息和功能分析,可以了解变异位点的可能功能和生物学影响,并推测其可能的疾病相关性。
- 突变负担和遗传背景:通过对样本中其他突变位点的分析,可以了解突变负担和遗传背景对变异位点的影响。
- 组合变异和复杂遗传模式:对于一些复杂的遗传疾病或多基因遗传模式,需要结合多个变异位点进行分析,以确定它们之间的关系和可能的影响。
总之,基因测序数据的解读和分析是一个复杂而重要的过程,需要综合运用多种工具和方法,从不同的角度进行分析,以获得有用的生物学信息。在实际应用中,需要根据具体情况选择合适的技术和策略,以便更好地解读和分析基因测序数据。 |
|

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 






 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 发表于 2025-2-27 05:44
发表于 2025-2-27 05:44