金桔
金币
威望
贡献
回帖0
精华
在线时间 小时
|
登陆有奖并可浏览互动!
您需要 登录 才可以下载或查看,没有账号?立即注册


×
目录
1. 概述
2. 一代测序技术
3. 二代测序技术
4. 三代测序技术
5. 四代测序技术
6. 各代测序技术比较
7. 基因测序的应用领域
8. 一代测序注意问题
9. 二代测序相关的名词解释
10. 三代全长转录本分析工具
1. 概述
基因测序技术(DNA sequencing)是指获得目标DNA片段碱基(包括腺嘌呤A、胸腺嘧啶T、胞嘧啶C、鸟嘌呤G)排列顺序的技术。在基础生物学研究,以及包括医学诊断、生物技术开发、法医生物学、系统生物学、微生物等不断拓展的多个其他应用领域中,基因测序技术已成为极其重要的专业技术之一,现在基因测序技术已经能帮助科学家获得人类基因组以及其他许多动植物和微生物物种的完整DNA序列。

从20世纪70年代到现在有很多测序技术和平台的产生,其中包括SBC法、454、Ion Torrent、SBL法、Sanger法、Illumina、Pacbio、Nanopore等。近半世纪以来,基因测序技术飞速发展,从一代发展至四代,发生了日新月异的变化。
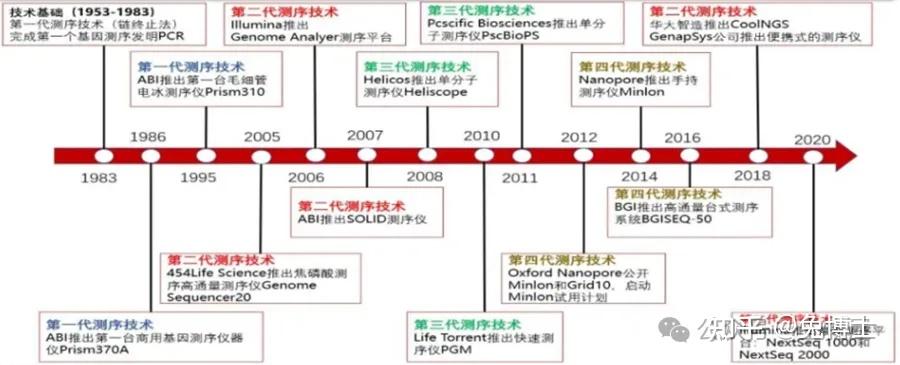
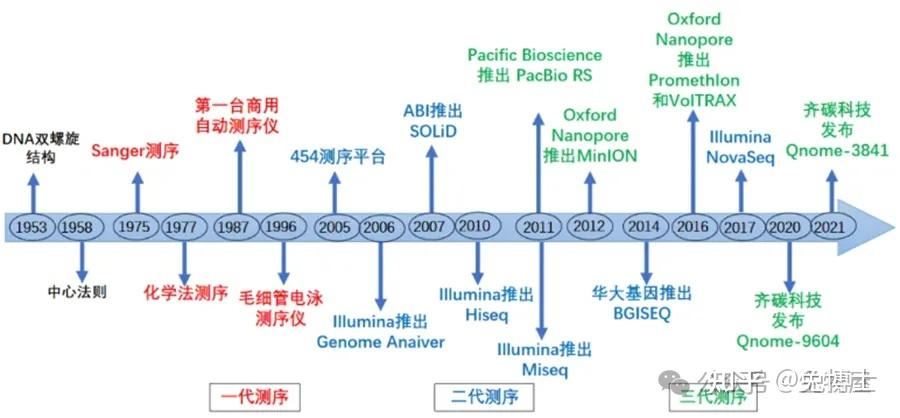
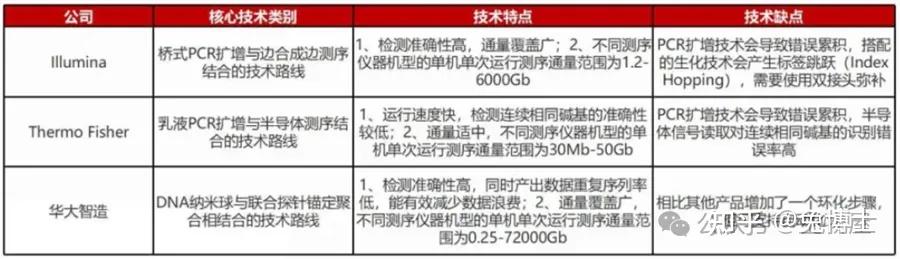
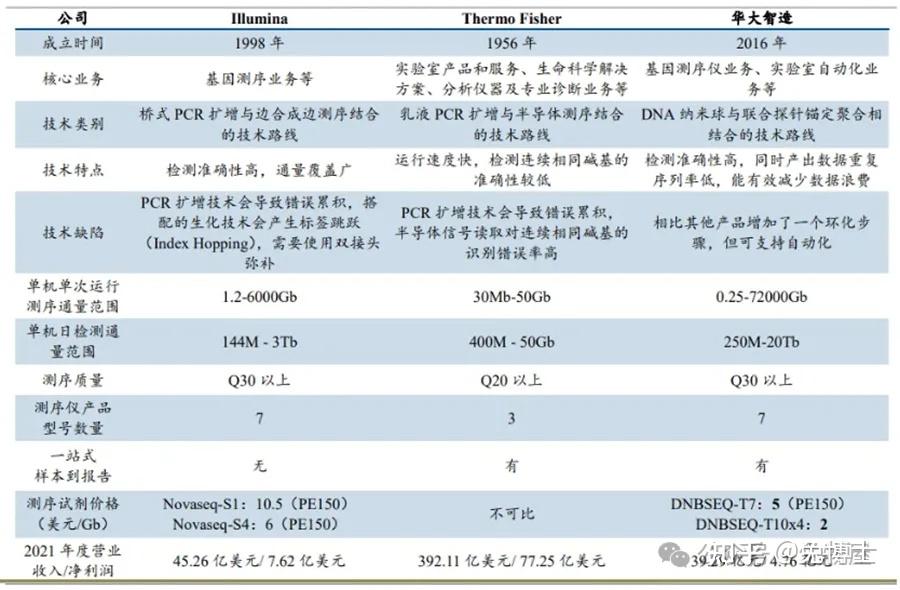
第一个人类的基因组,从1990年到2003年,由2000名科学家历时13年,花费38亿美金才完成,图谱中包含了人类染色体的近30亿个碱基对的核苷酸序列,由于高度重复的DNA块组成,当时技术的局限,这份图谱仍留下了约8%的空白区,这部分的测序难度非常大。1975年至今,基因测序技术已经发展到第四代,测序时间从13年缩短到5小时,测序金额从38亿美金降低到1000元人民币。基因测序技术的核心是基因测序仪的核心,作为基因测序产业链上游的进入壁垒最高技术之一,全球具有自主研发并量产临床级高通量基因测序仪能力的企业主要有Illumina、Thermo Fisher及华大智造。
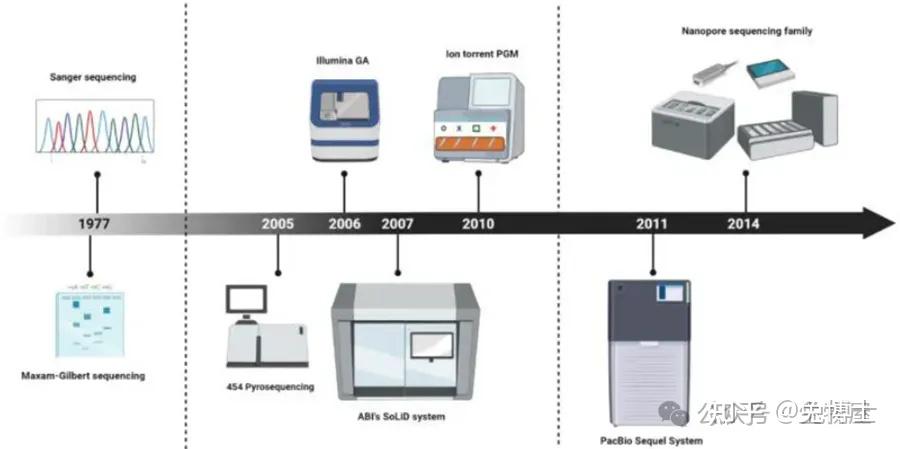
2. 一代测序技术
1975年,英国化学家桑格(Frederick Sanger,1918-2013)发明了双脱氧链终止法,该技术使用双脱氧核苷酸,它在复制过程中终止DNA 链的链伸长,并允许产生长度高达数百个核苷酸的序列读数。Sanger的方法被广泛采用,并通过实现 DNA 和 RNA 的快速测序而彻底改变了分子生物学领域。1976-1977年,吉尔伯特(Walter Gilbert,1932-至今)和他的学生Allan Maxam(1942-至今)发明了化学降解法,可以对完整噬菌体 PhiX174 进行测序。这两种测序方法都被称为一代测序技术。1987年,第一台商业自动化测序仪Applied Biosystems ABI 370在美国推出,该机器利用荧光标记的双脱氧核苷酸和毛细管电泳实现Sanger测序方法的自动化,显著提高了DNA测序的速度和准确性。ABI 370 迅速成为行业标准,随后技术的改进导致了能够产生更长读取的更高通量测序仪的开发。虽然第一代技术已基本上被更新的、更高通量的测序技术所取代,但它仍然是测序技术发展的重要历史里程碑。
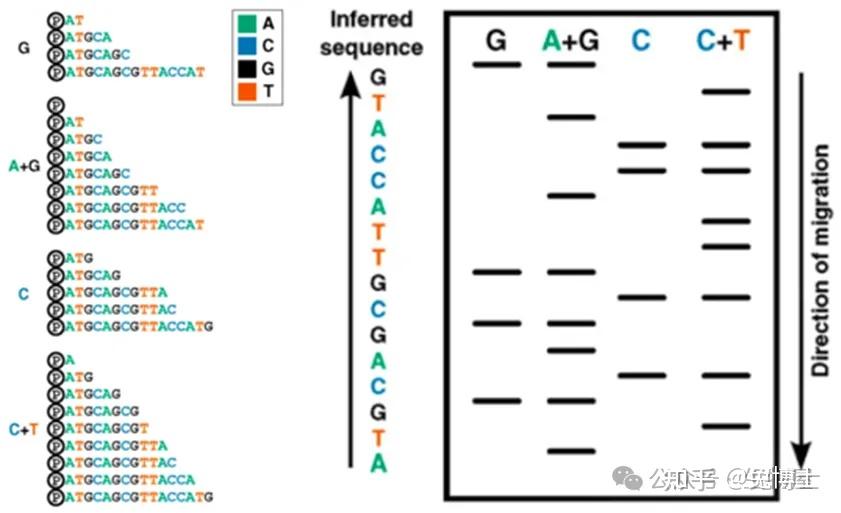
(1)化学降解法:即Maxam-Gilbert化学降解法原理:将DNA片段的5’端磷酸基使用放射性同位素标记,再分别采用不同的化学试剂处理修饰和裂解特定碱基,从而产生一系列长度不一而5’端被标记的DNA片段,这些以特定碱基结尾的片段群通过聚丙烯酰胺凝胶电泳分离,再经放射线自显影,确定各片段末端碱基,从而得出目的DNA的碱基序列。
(2)双脱氧链终止法:即Sanger法原理:先合成DNA,再分析测序结果。由于双脱氧核苷酸(ddNTP)的3’位置脱氧,其在DNA的合成过程中不能形成磷酸二酯键,因此可以用来中断DNA合成反应,在4个DNA合成反应体系中分别加入一定比例带有放射性同位素标记的ddNTP(分为:ddATP、ddCTP、ddGTP和ddTTP),通过凝胶电泳和放射自显影,根据电泳带的位置确定待测分子的DNA序列。在每个反应体系中,ddNTP相对于dNTP是很少的,所以只有部分新链在不同的位置特异性终止,最终就会得到一系列长度不一的序列。Sanger法测序读长长、准确度高,但是通量不高。总的来说,第一代测序技术的主要特点是测序读长可达1000bp,准确性高达99.999%,但其测序成本偏高(相对基因组大小),通量低等方面的缺点,严重影响了其真正大规模的应用。因而第一代测序技术并不是理想的测序方法。优点:一代测序准确性最高,几乎达到100%,是公认的测序技术的“金标准”。缺点:一代测序的缺点是速度慢、费用高、只能同时测序一条DNA模板。费用:3000元左右。
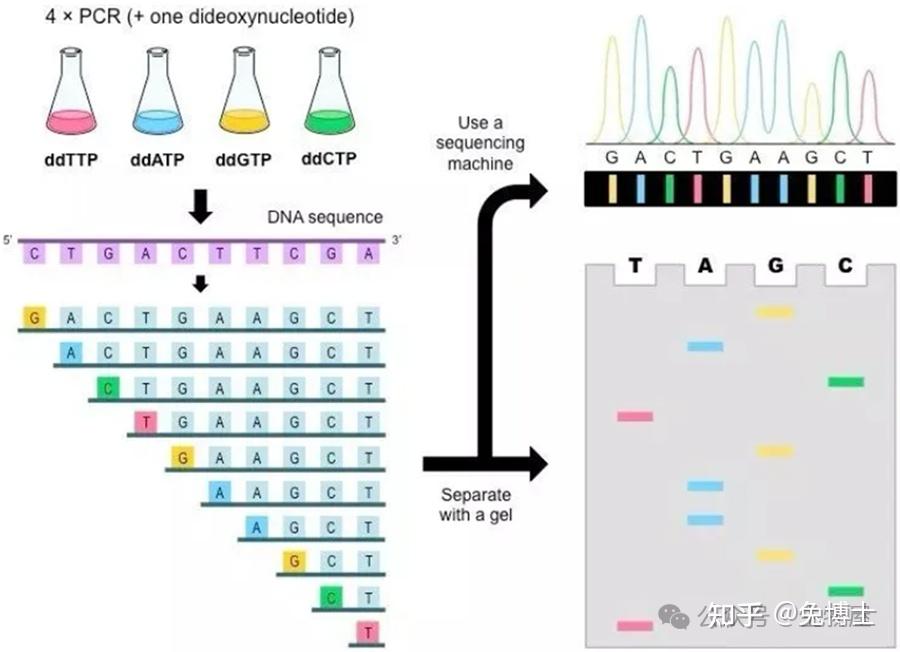
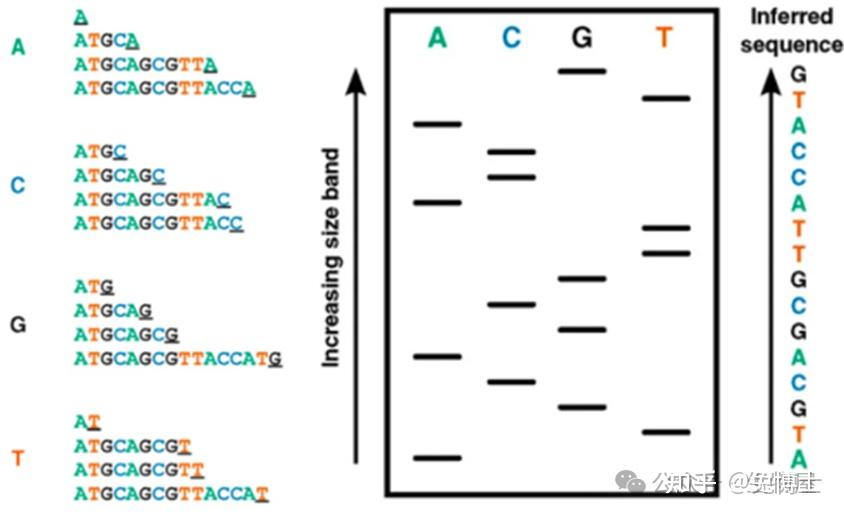
测序过程:在一个反应体系中,加入很多相同的DNA模板链、引物、dNTPs、ddNTPs、DNA合成酶。
引物,简单来说是互补链的“引路者”;
dNTPs是DNA合成所需的原料;
ddNTPs (ddATP、ddGTP、ddCTP、ddTTP) 也是DNA合成的原料,同时是DNA合成的“终结者”;
DNA合成酶帮助底物(dNTP或ddNTP)连接在正在合成的DNA链上。随着反应的进行,ddNTP这个“终结者”参与的位置不同,终止的位置也不同,因此合成的DNA长短不一,分子量也就不同。
测序时,在PCR扩增、PAGE分离DNA后,再借助显微镜,就可以得到PAGE条带,一个条带代表一种分子量的DNA。DNA带负电,可以从负极跑到正极,分子量越小的DNA跑得越快,跑得越快的条带越先合成,就这样按顺序读取对应的反应体系,就可以得到整个DNA的核苷酸顺序。
要区分“终结者”的类型,除了构建四个反应体系,还可以借助荧光素:让不同的ddNTPs显示不同的颜色,最终用荧光检测仪检测,读取后就可以得到测序结果了。不仅如此,人们还想到用不同发射波长的荧光基团来标记ddNTPs,在激光的照射下显示不同的颜色,这种光波长信号可以直接用计算机来识别。
3. 二代测序技术
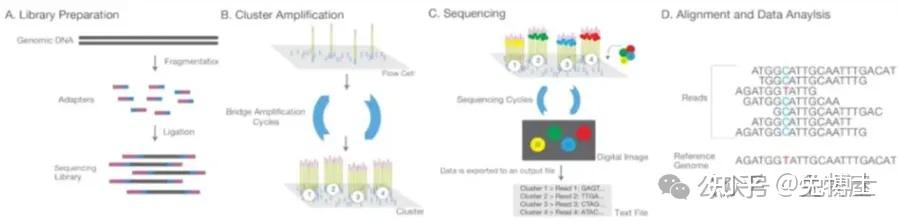
二代测序技术也称下一代测序(Next generation sequencing, NGS)能够同时对数千至数百万个DNA 片段进行测序,从而彻底改变了DNA 测序技术,这些方法与传统Sanger测序的不同之处在于它们执行并行测序的能力。二代测序技术在大幅提高了测序速度的同时,还大大地降低了测序成本,其测序成本近五年来从几千元1G(1G即10亿碱基)降到了到今天的40多块钱1G数据量,并且保持了高准确性,以前完成一个人类基因组的测序需要3年时间,而使用二代测序技术则仅仅需要1周,但其序列读长方面比起第一代测序技术则要短很多,大多只100bp-150bp。
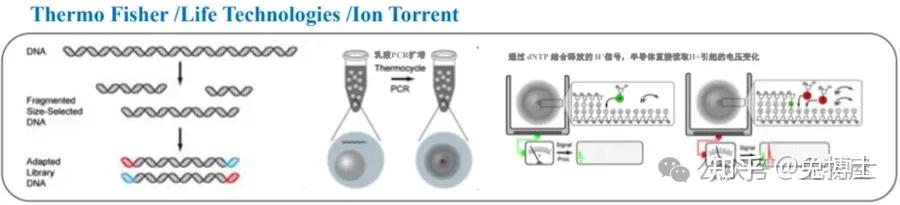
二代测序技术基于大规模平行测序技术(massive parallel analysis,MPS)方法对大量的目的基因同时进行测序,二代测序平台主要采用的技术有以下三种:边合成边测序(sequencing by synthesis,SBS),连接法测序(sequencing by ligation,SBL,又名SOLiD)和半导体测序(Ion Torrent)。罗氏的454测序方法依赖于焦磷酸测序,通过检测将核苷酸添加到DNA模板时焦磷酸盐的释放来确定序列;Ion Torrent 测序是通过检测 DNA 合成过程中氢离子的释放来确定序列;广泛使用的Illumina测序平台采用基于可逆染料终止子的合成测序方法;SOLiD测序(寡核苷酸连接和检测测序)采用基于连接的方法,使用可逆终止子来确定 DNA 序列。
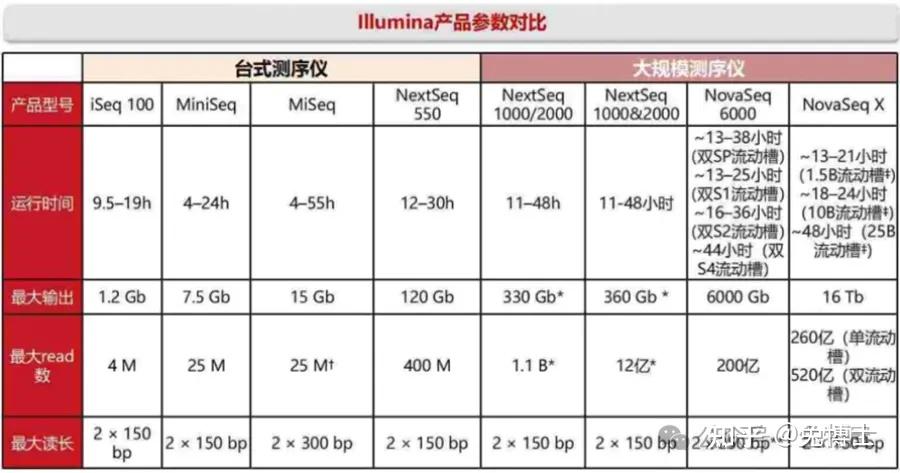
目前illumina的测序仪占全球75%以上,以NextSeq、HiSeq、NovaSeq等系列为主,它的机器采用的都是边合成边测序的方法,读长短(50-300bp),准确度达99.9%,通量很高。以Illumina平台为代表的第二代测序技术实现了高通量测序,有了革命性进展,使得大规模并行测序成为现实,极大推动了生命科学领域基因组学的发展。Illumina循环SBS法(cycle SBS)即SBRT(Sequencing By Reversible Termination,可逆终止)的核心技术是DNA合成的可逆性末端循环,即3'-OH可逆性的修饰和去修饰。
常见测序技术与平台
2005年,454生命科学公司(454 Life Sciences)推出了革命性的基于焦磷酸测序法的超高通量基因组测序系统——Genome Sequencer 20 System,开创了边合成边测序的先河。
2007年初,罗氏诊断(Roche Diagnostics)收购454公司。Roche 454测序系统是第一个商业化运营二代测序技术的平台。
2013年,罗氏诊断宣布关闭454测序业务,并于2016年终止相关服务。
- Illumina公司 Solexa/HiSeq/MiSeq/NextSeq测序
Illumina公司的二代测序仪应该说是目前全球使用量最大的第二代测序机器。
SoLid测序技术是ABI公司于2007年开始投入用于商业测序应用的仪器,目前该平台已淡出市场。
- 华大基因 Complete Genomics/DNBSEQ测序
华大基因于2013年收购美国Complete Genomics(CG)公司,获取了其测序核心技术和其他核心知识产权,随后自己组建研发团队,不断增强自身实力。
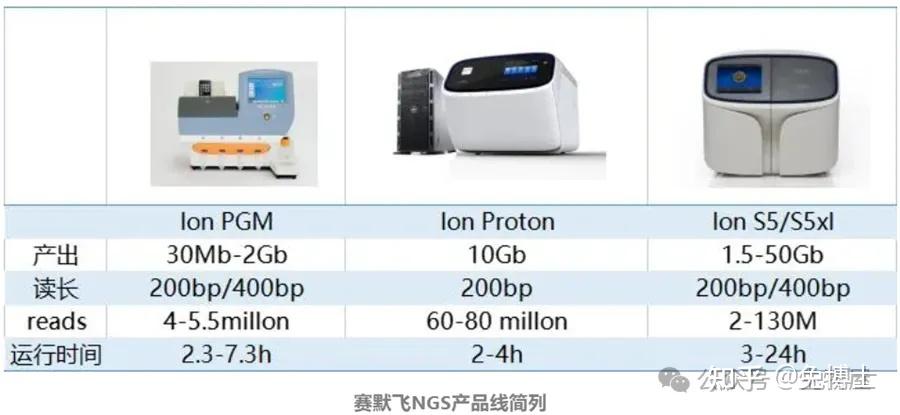
2007年,454创始人罗森伯格创办Ion Torrent公司。
2010年,Ion Torrent推出了世界上第一台半导体测序仪——个人染色体检测仪PGM。
2010年,Life Technologies收购了Ion Torrent。
2012年,Life公司再接再厉推出了功能更为强大的Ion Proton测序仪。
2013年,赛默飞世尔科技公司(Thermo Fisher Scientific)收购Life Tech。
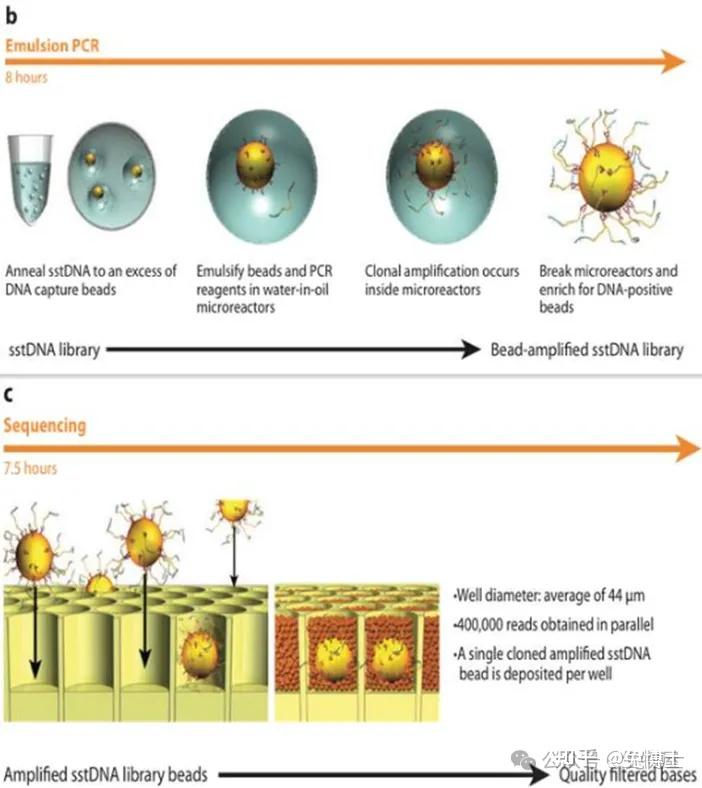
(1) Roche 454焦磷酸测序:使用边合成边测序技术,避免了Sanger法存在的宿主菌克隆问题。即首先将目的DNA片段打断成300-800bp的小片段,然后在5’端加上一个磷酸基团,并将3’端变成平端,再在两端加上衔接子组成目的DNA的样品文库。
之后将目的DNA片段固定到磁珠上,将磁珠包被在单个油水混合小滴中进行独立的扩增,从而实现所有目的DNA片段进行平行扩增PCR。
随后将这些DNA放入PTP反应板中共进行后继测序,这里面包含了化学光反应所需的各种酶和底物。
测序开始时,将T、A、G、C按顺序循环单分子进入PTP 板,如果发生配对,则会释放一个焦磷酸盐分子,其在后续与ATP磷酸化酶和虫荧光素反应产生光信号,此光信号被捕获以确定碱基序列。
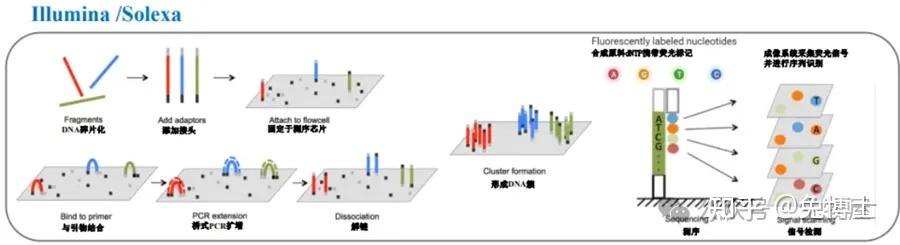
(2)Illumina Solexa合成测序:桥式PCR+4色荧光可逆终止+激光扫描成像,使用克隆单分子阵列技术。
首先将目的DNA片段打断成100-200bp,随机连接到固相基质上,经过Bst聚合酶延伸和甲酸胺变性的桥式PCR循环,生成大量的DNA簇。
之后的反应与Sanger法类似,每次延伸所产生的光信号被标准的阵列光学检测系统分析测序,下一次循环中把终止剂和荧光标记基团裂解掉,然后继续延伸dNTP,实现了边合成边测序技术。
基本原理:边合成边测序。将dNTP的3'-OH以叠氮基团RTG(Reversible Terminating Group,可逆末端基团)进行修饰;将4种碱基分别与不同的荧光分子连接;DNA合成时,RTG能起到类似于ddNTP的作用终止反应;每次合成反应终止并读取信号之后,洗脱RTG和荧光分子,进行下一轮循环。
优点:二代测序高通量,非常适合进行基因组、转录组以及表观遗传学方面的检测。除此以外,单条序列测序成本非常低廉,仅仅相当于第一代测序技术的1%。
缺点:二代测序检测序列较短,测序前需要PCR扩增,错误率比一代稍高,为了降低错误率,可以使用Sanger测序技术对第二代测序技术检测出的变异进行验证,这也正是Sanger测序沿用至今的原因。
费用:5000-8000元左右。
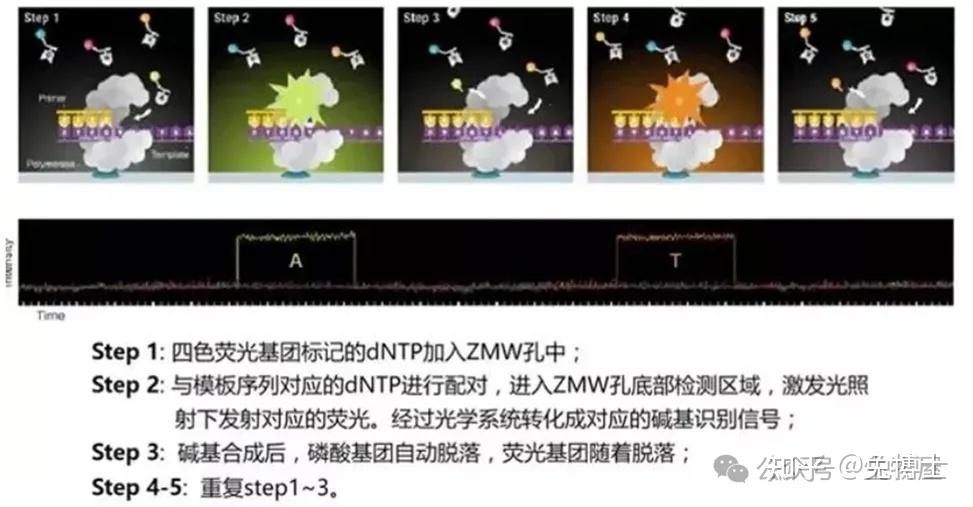
测序过程:
① DNA文库制备:超声打断加接头;
② Flowcell:吸附流动DNA片段;
③桥式PCR扩增与变性:放大信号;
④测序:测序碱基转化为光学信号。
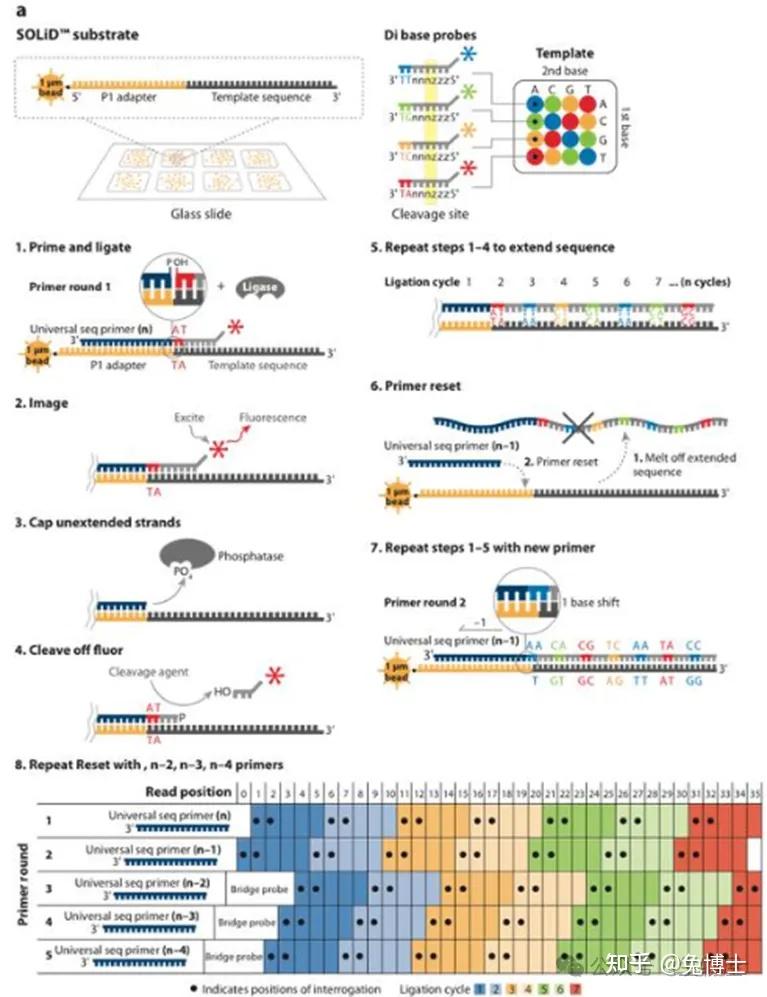
(3)ABI SOLiD连接法测序:首先制备DNA文库,可以使用片段文库和配对末端文库。第二阶段与焦磷酸测序相同,加入磁珠等反应元件进行乳液PCR(emPCR)平行扩增,不同的是该方法的磁珠只有1 µm。
在连接测序中,底物是8 个碱基的八聚体单链荧光探针,在5′末端分别标记了CY5、Teaxs Red、CY3、6-FAM这四种颜色的荧光染料。3′ 端的第 1、2 位碱基类别排序分别对应着一个固定的荧光染料,第 3、4、5 位碱基“n”是随机碱基,第 6、7、8 位碱基“z”是可以和任何碱基配对的特殊碱基。一次测序中包括了五轮连接反应,可以减小测序误差。
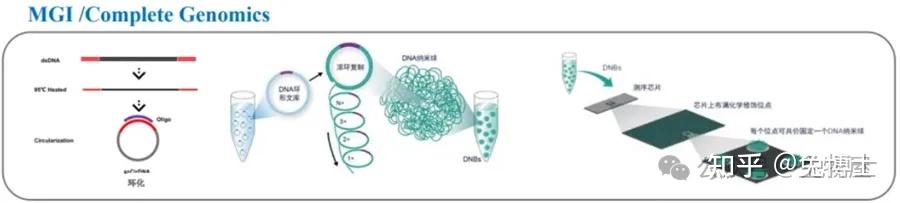
(4)华大基因Complete Genomics测序:DNA 纳米球+双色测序+联合探针锚定聚合
华大智造测序仪采用先进的DNBSEQ测序核心技术,通过仪器气液系统先将DNA纳米球(DNA nanoball,DNB)泵入到规则阵列芯片(Patterned Array)并加以固定,然后泵入测序模板及测序试剂。
测序模板与芯片上的DNB 的接头互补杂交,在DNA聚合酶的催化下,测序模板与测序试剂中的带荧光标记的探针相结合。
然后由激光器激发荧光基团发光,不同荧光基团所发射的光信号被相机采集,经过处理后转换成数字信号,传输到计算机进行处理,可以获取待测样本的碱基序列信息。
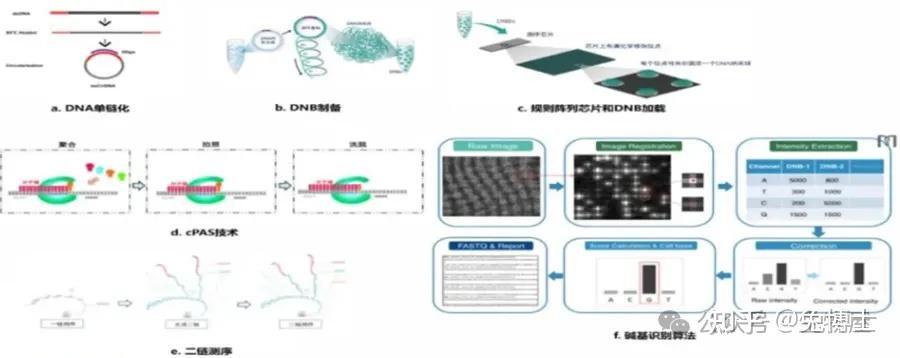
测序过程:
① DNA 文库制备:DNA 碎片化后加接头,环化得到单链环状 DNA;
②滚环扩增:无损复制并形成 DNA 纳米球;
③规则阵列芯片:每个结合位点固定一个DNA纳米球;
④测序:DNA纳米球与酶和荧光探针反应,采集和识别荧光信号。
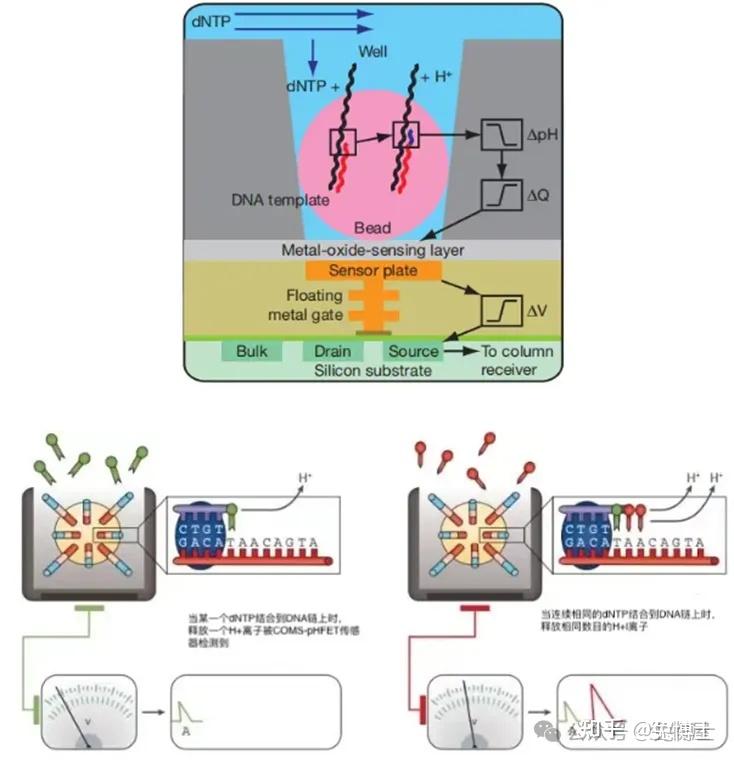
(5)2.5代测序技术/半导体测序技术:半导体测序技术由Ion Torrent研制开发,乳液PCR +4种dNTP +微电极pH检测。由于使用到了Emusion PCR技术,其实质介于二代和三代测序技术之间。该技术使用一种高密度半导体芯片,每个芯片单独的进行测序。实验时先将芯片置于一个离子敏感层和离子感受器之上,当DNA聚合酶在每一个单分子模板链上滑动时,发生聚合反应,释放出氢离子,最终离子感受器就会捕捉到这种信号,从而读出DNA序列。
测序过程:
① DNA文库制备:喷雾打断加接头;
②乳液PCR:注水入油独立PCR;
③微电极pH检测:磁珠入池记录pH。
4. 三代测序技术
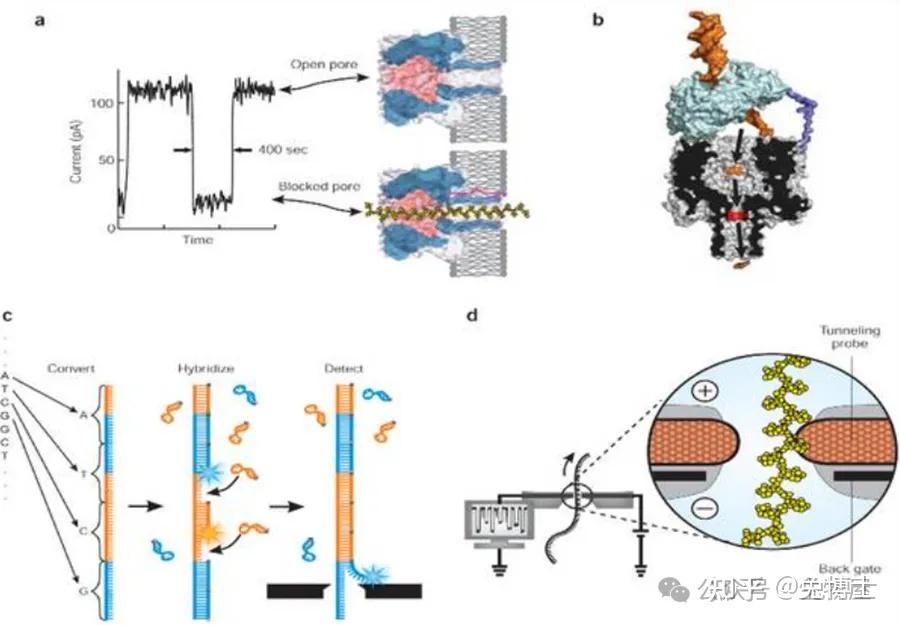
三代测序技术代表了DNA 测序的最新进展,提供了克服前几代测序局限性的新方法,这些技术提供长读长测序功能,与早期方法相比,能够对更大的DNA 片段进行测序。单分子荧光测序技术代表是美国螺旋生物(Helicos BioSciences)(该司已停止营业)的SMS技术和美国太平洋生物(Pacific Bioscience)的SMRT(Single Molecule Real-Time Sequencing)技术。PacBio开发的SMRT测序技术建立在两项重要的发明基础之上,从而攻克了测序领域测序读长短的重大难题。第一,零模波导孔技术(Zero-mode Waveguides,ZMWs)使激发光被限定在单分子纳米孔底部一定范围内,过滤了背景噪音;第二,荧光基团结合在核苷酸的磷酸基团上,帮助DNA聚合酶完成一个全天然的DNA链合成过程。与前两代相比,最大的特点就是单分子测序,测序过程无需进行PCR扩增,超长读长,是二代测序技术的100倍以上。这种纳米孔单分子测序仪还有另一大特点,它能够直接读取出甲基化的胞嘧啶,而不必像二代测序方法那样需要事先对基因组进行bisulfite处理。
基本原理:Pacbio仍然采用边合成边测序的原理,其通过将脱氧核苷酸用荧光标记,实时地记录荧光的强度变化。当荧光基团被掺入DNA链的时候,它的荧光就同时能在DNA链上探测到。当它与DNA链形成化学键的时候,它的荧光基团就被DNA聚合酶切除,荧光消失。这种荧光标记的脱氧核苷酸不会影响DNA聚合酶的活性,并且在荧光被切除之后,合成的DNA链和天然的DNA链完全一样。测序过程包括文库构建和上机两步。文库构建是将长片段DNA分子与测序接头连接成茎环结构,然后加上与接头互补的测序引物及DNA聚合酶。上机测序是将构建好的文库复合物载入SMRT Cell的纳米孔中,通常一个纳米孔固定一个DNA分子,DNA聚合酶通过共价连接的方式固定在纳米孔底部。
基于该原理的具体产品有PacBio Sequel测序仪、PacBio Sequel II测序仪,PacBio Sequel测序仪是首个商业化应用的第三代测序技术平台,其打破传统短读长测序诸多技术瓶颈。PacBio Sequel II测序仪是PacBio Sequel的升级款,可提供CLR Library和CCS library(HIFI)两种测序模式,测序芯片上的导孔(ZMW)由100万个提升至800万个,理论通量提升8倍。CCS reads单碱基准确性有了极大提升,同一片段测序4次后,单一read的准确性可达99%。
优点:Pacbio三代测序设备在DNA序列片段读长上优于二代设备,测序读长长、通量高、可进行甲基化的直接测序。
缺点:在准确度上较二代设备差,准确度不高,但可通过测序深度弥补,GC偏差低,
费用:单样本的测序成本一直居高不下。
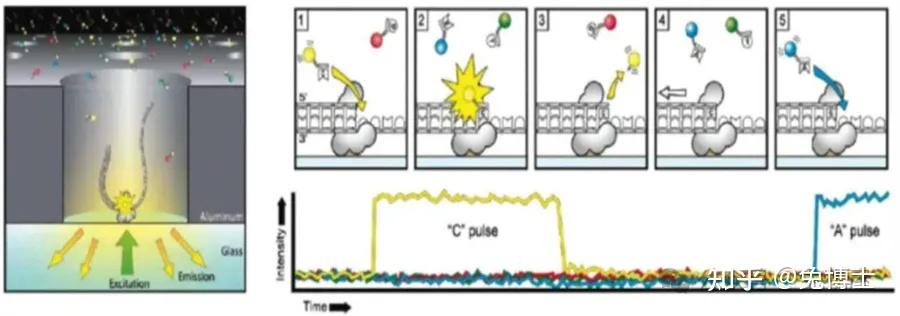
测序过程:
(1) 样本的准备:将提取的DNA或RNA,通过接头序列直接构成亚玲状或者圆环状。
(2) 合成与测序:通过测序芯片测序。测序芯片含大约15万个直径为70nm的测序微孔,为反应提供很好的场所,并且每一个小孔都带有链酶亲和素。在反应体系中加入所需的样本、引物、dNTPs(带有荧光基团)、DNA聚合酶。但这里的DNA聚合酶会稍有不同--带有生物素。生物素与链酶亲和素具有极强的的亲和力,所以DNA聚合酶就被固定在了测序微孔里。这样,只有参与合成的dNTP发出的激光才会被捕捉到,通过强大计算机分析芯片里的光谱就得到测序结果。
5. 四代测序技术
以英国牛津纳米孔(Oxford Nanopore Technologies,ONT)公司为代表的纳米孔测序技术与其他测序技术不同的是,它基于电信号而不是光信号。经历了三个主要的技术革新:一、单分子DNA从纳米孔通过;二、纳米孔上的酶对于测序分子在单核苷酸精度的控制;三、单核苷酸的测序精度控制,主要是通过ssDNA或RNA模板分子通过纳米孔而带来的“电信号”变化推测碱基组成进行实时测序。
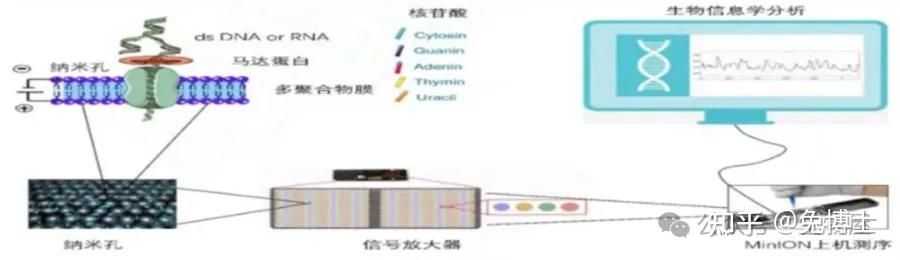
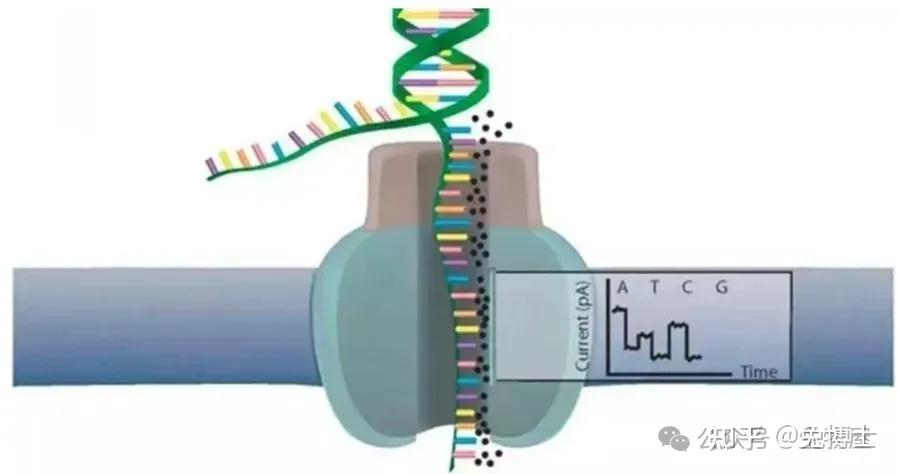
基本原理:将在某一面上含有一对电极的特殊脂质双分子层置于一个微孔之上,该双分子层中含有很多由α-溶血素蛋白组成的纳米孔(直径2.6nm),只能容纳一个核苷酸通过,并且每个纳米孔会结合一个核酸外切酶。当DNA模板进入孔道时,孔道中的核酸外切酶会“抓住”DNA分子,顺序剪切掉穿过纳米孔道的DNA碱基,每一个碱基通过纳米孔时都会产生一个阻断,根据阻断电流的变化就能检测出相应碱基的种类,从而进行实时测序,最终得到DNA分子的序列。由于其原理与其他平台有较大差异,亦有被称为第3.5代或四代测序技术。Nanopore测序仪的具体产品种类很多,均基于Nanopore芯片来搭建的平台,大到由多个芯片阵列组成的PromehION、GridION系列测序仪,小到可以连接手机的Type C、电脑USB的MnION系列便携式测序仪。其中PromethION是一款高通量、高样本数的台式系统,基于模块化设计(多达 48 个测序芯片,各有多达 3000 个纳米孔通道,总计达 144000 个),测序芯片既可单独也可同时运行,尤其适合于大样本量、具有庞大数据量的项目。
优点:是单分子测序,通量高、测序读长长(超过150kb),测序速度快,测序数据实时监控,机器方便携带等。
缺点:单芯片测序成本还是在几百美金以上,准确度低,不可通过测序深度弥补,但可通过Illumina read 纠错。
主要特点:
1) 超长读长:在纳米孔测序中,读长长度不受限于测序设备,可以通过所使用的文库制备实验方案来控制片段长度。目前DNA片段长度最高记录为900kb。
2) 读取速度快:MinION流动单元每秒可读取500bp。
3) 直接测序:纳米孔技术基于电子学原理,允许直接测序原始DNA和RNA。不需要通过DNA拷贝、进行链合成,节省了时间和成本。由于纳米孔技术支持无需PCR的直接测序,也就没有了扩增偏好性,并且文库制备工作流程也更简单。
4) 通量高:PromethION包含48个独立流动单元,最多可以在2天内输出2-4TB的数据量
5) 便携:ONT MinION只有USB设备大小,又称为掌上测序仪,在电脑上即可对数据进行读取。
但同时由于该技术拥有超过1000 种独立的信号,其错误率也较高(主要表现为对 Indel 的检测)。
由于修饰的碱基会改变原有设定的电压变化,所以碱基的修饰对 ONT 而言同样是一大挑战。
测序过程:
1) DNA双链解螺旋,解开成为单链DNA。同时DNA解旋酶也作为马达蛋白促使DNA单链分子通过一个以α-溶血素来构建的生物纳米孔,孔道内表面覆盖有一种合成的环糊精作为转换器。
2) DNA单链停留在孔道中,与孔内的环糊精短暂地相互作用,影响了流过纳米孔原本的电流,带来了电流变化。而不同的碱基带来的电流变化不同,例如A与T的电信号大小很接近,但T在环糊精停留的时间是其他核苷酸的2-3倍,因此每个碱基都因其产生电流干扰振幅是特有的而被区分开来。
3) 根据电流变化的频谱,应用模式识别算法得到碱基序列。
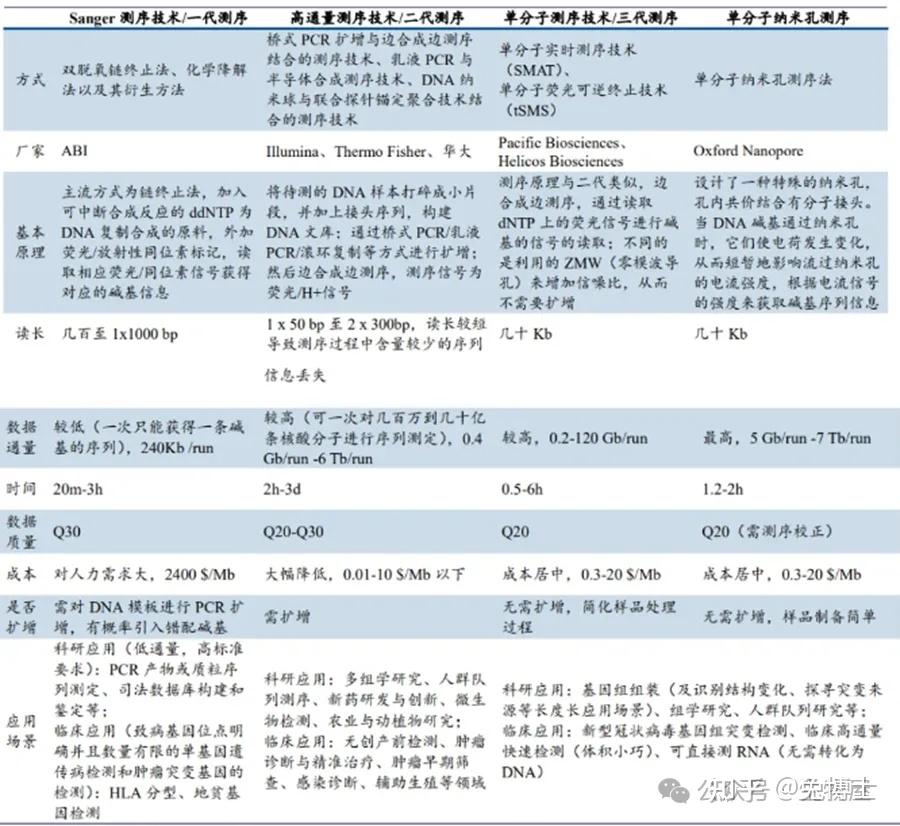
6. 各代测序技术比较
各代测序技术的优缺点汇总
优点:方法简便,分辨率高,测序片段长,流程细致,质控环节多,污染低,结果直观可视,假性结果极低。在单个基因测序方面具有优势。金标准。
缺点:测序试剂昂贵,通量低,处理比较长的同聚物时也有自身的问题。
优点:通量高,一次可对几十万到几百万条核酸分子进行序列测定;单条序列成本低。缺点:序列读长短;建库过程中有PCR过程,会引入错配碱基;要想得到准确和长度较长的拼接结果,需要较高的测序覆盖率,会增加成本和错误率。
优点:1) 连贯性:连贯性对基因组的组装非常重要,如果连贯性比较好,能够准确的反应出基因结构之间的关系(外显子、基因簇、转移元件、调节序列等)。2) 完整性:如果一个基因组的测序深度>50X,理论上每一个碱基都会被测到。但实际上,基因组仍然会有很多缺失区域,比如即便是最新的人类参考基因组,其中仍然会有超过百万的“N”。读长的提升能够有效提高基因组组装的完整性。3) 准确性:基因组组装的准确性可以在核酸水平或者结构变异水平进行描述。Illumina的三代测序技术的准确性非常高,每个碱基准确性大于99.9%,PacBio和Nanopore的准确性在足够测序深度的情况下,经过算法校正之后也能够达到99.9%。
缺点:单读长的错误率偏高,需要重复测序以纠错;依赖DNA聚合酶的活性;成本较高;生信分析软件不够丰富,数据积累少。
优点:1) 超高的便携性,MinION可以做到即插即用,非常适合流行病疫区的即时检测;2) 超长的读长,MinION可以达到150kb,可用来填补之前测序的gap,可用于从头组装基因组,可变剪切等;3) 广泛的应用,可以直接对DNA,RNA和蛋白质序列进行测序;碱基判读准确率较高,R10纳米孔数据质量值超过Q40(即错误识别的概率是0.01%,即错误率0.01%),一致性(Identity)质量值达Q50 (99.999%的碱基准确率)。
缺点:低准确度,尤其是2D reads,虽然可以通过降低DNA通过速度或一致性检测提高准确度,但仍无法和二代测序相比。
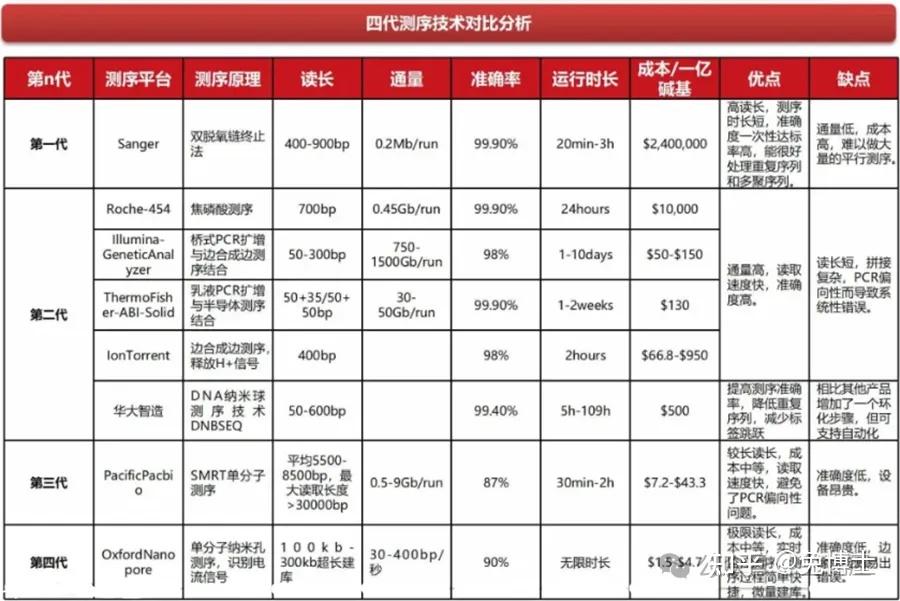
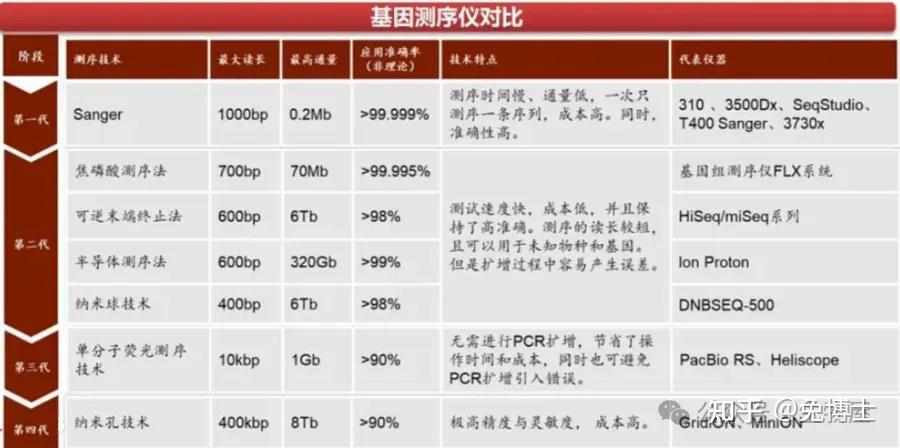
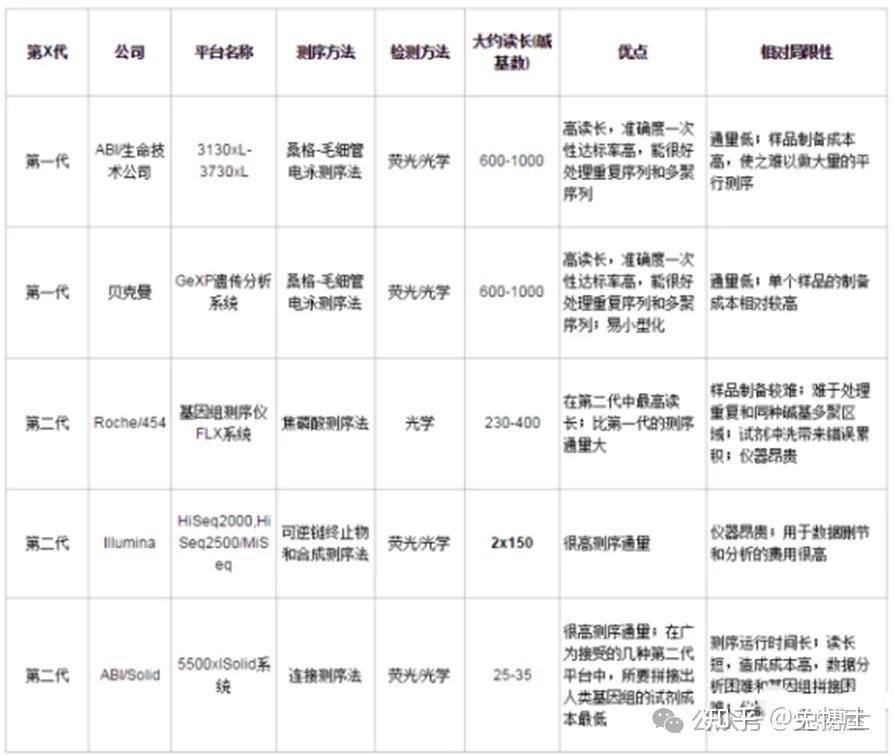
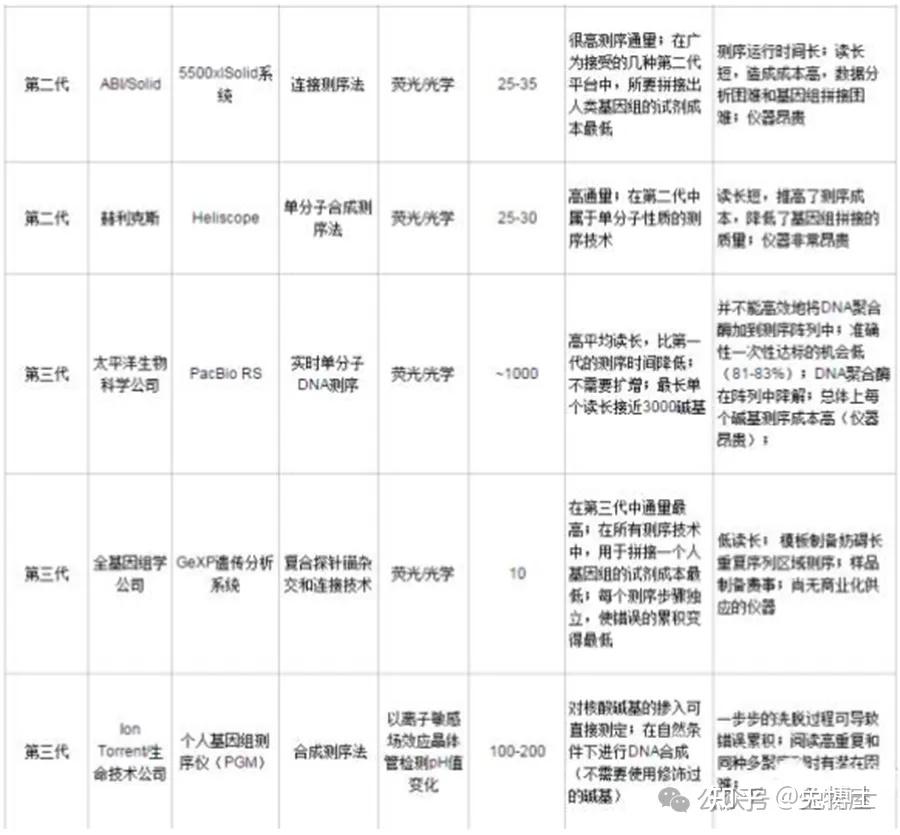
NGS测序比较在NGS测序方向主流技术主要分3类:联合探针锚定聚合测序法(华大)、边合成边测序法(因美纳)、半导体测序法(赛默飞)。其中Sanger测序逐渐被NGS挤压,然后应用场景萎缩,例如法医诊断等等;三代测序/单分子测序目前主要应用场景主要是科研研究领域。


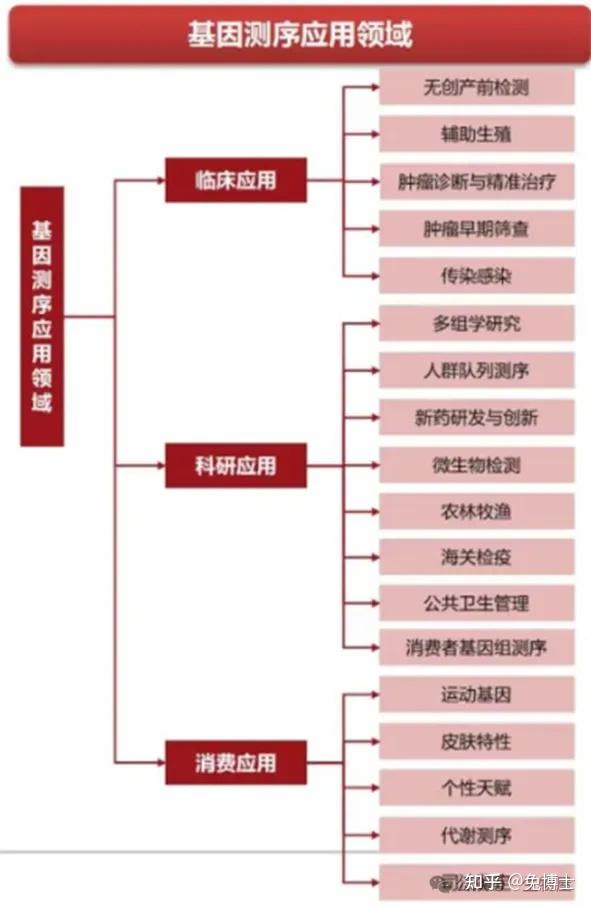
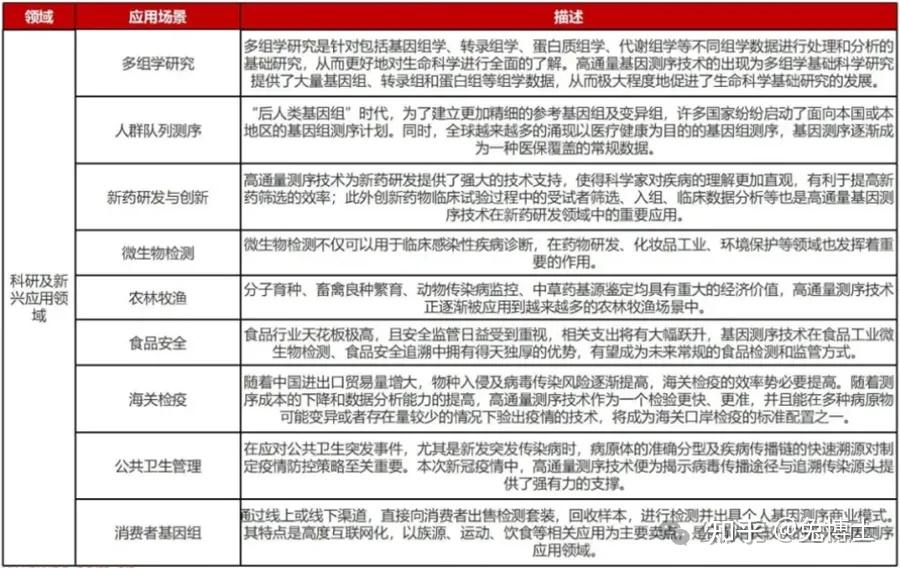
7. 基因测序的应用领域
基因测序技术的迅猛发展,不仅推动了生命科学的研究,更在多个领域中展现出了其独特的价值和广泛的应用前景。
在医学领域,基因测序技术已经成为了诊断遗传性疾病的重要手段。通过分析患者的基因组,医生可以识别出导致疾病的特定基因变异,从而提供更为精确的诊断信息。此外,基因测序还为个性化医疗的实现提供了可能。根据个体的基因信息,医生可以为患者定制个性化的治疗方案,选择最合适的药物和治疗方法,以达到最佳的治疗效果。
在农业科学中,基因测序技术的应用同样广泛。科学家通过测序作物的基因组,可以发现影响作物产量和品质的关键基因。这些信息对于作物的遗传改良和育种至关重要。通过基因编辑技术,可以培育出更加健康、抗病虫害、适应性更强的新品种,从而提高农业生产的效率和可持续性。
法医学中的身份鉴定和亲子鉴定也离不开基因测序技术,通过比对DNA样本,法医专家可以确定个体之间的亲缘关系,或者在犯罪现场找到关键的遗传证据,这些技术在司法案件的侦破和判决中起着至关重要的作用。
基因测序技术还被广泛应用于生物多样性的研究中,通过测序不同物种的基因组,科学家可以探索它们之间的进化关系,理解生物多样性的形成和演变,这些研究对于保护濒危物种、维护生态平衡具有重要意义。

8. 一代测序注意问题
1) 测序结果不到800Bases是什么原因?(1)G/C rich、G/C Cluster。这种情况一般表现为测序信号突然减弱或消失如在DNA样品中的DNA序列分布匀称,没有复杂结构时,正常的测序反应能保证达到800Bases以上。但有一些DNA样品立体结构复杂,造成聚合酶延伸反应终止,测序信号突然减弱或消失,或者测序结果出现套峰现象,出现这些现象的原因由DNA模板本身所造成。
(2)A、T的Poly结构这种情况一般表现为A、T连续结构后面的测序结果出现套峰。根据文献记载。原因在于聚合酶进行聚合反应时,由于A或T的连续,聚合酶难以识别完整的每个A或T,在某个A或T的后面便开始进行A或T连续结构以后序列的聚合反应(打滑现象),造成测序结果紊乱,出现套峰。一般在多少个A或T的后面能出现这种情况呢?现在还没有这方面的报道。根据我们的经验,这一情况的出现和A或T的连续结构后面的序列的排列情况有着直接的关系。有时10多个A或T的连续结构后面便出现套峰,但有时60~70个A或T的连续结构后面的序列也一样可以完整地读出来。具体情况还有待考证。一般来说,PCR片段直接测序时,A或T的连续结构后面的序列测序结果都会出现套峰。原因在于测序时经历了PCR反应及测序反应(测序反应本身也是PCR反应)二次聚合酶的打滑现象。
(3)原因不明的复杂结构,测序结果出现突然信号减弱或消失从序列上看,DNA碱基排列并无特别异常。估计是DNA整体出现复杂结构,从某一位置开始聚合酶的聚合反应便无法进行。
2) 出现套峰是什么原因?
在测序反应中,模板或引物的原因都可能造成套峰的形成,归结其形成原因有以下几点:
(1)测序引物在模板上有两个结合位点;
(2)模板不纯,如果是质粒或是菌液,原因是非单克隆,如果是PCR,原因为非特异性条带;
(3)模板序列的特殊结构,如poly结构、发卡结构等;
(4)引物降解,或引物不纯。
套峰细分的话有如下几种情形:
①全双峰:多引物结合位点(针对菌液、质粒样品),非特异性扩增(针对PCR产物);
②前双峰:多引物结合位点,其中一套模板测序中断(针对菌液、质粒样品),多引物结合位点(PCR未纯化样品),引物二聚体或小片段干扰(针对PCR已纯化样品);
③中间双峰:非单克隆(针对质粒、菌液样品),碱基缺失或等位基因双模板(针对PCR未纯化样品);
④后双峰:非单克隆(针对菌液、质粒样品),碱基缺失(针对PCR样品);针对二聚体及小片段干扰的情况建议电泳切胶回收纯化;针对多引物结合位点的情况建议更换引物测序或反方向测通样品;针对碱基缺失建议克隆测序;针对非单克隆建议在克隆无误的前提下重新挑取单克隆测序;针对非特异性扩增建议优化反应条件重新制备样品测序;针对等位基因双模板建议克隆测序。
3) 样品测序无信号
可能是引物结合位点不存在或被破坏;建议更换引物测序或重新提供样品测序。
4) 样品测序信号差
可能是引物或模板的质量不高或是引物和模板的匹配性不好引起的,也可能是样品浓度偏低;建议提供高质量样品测序。
5) 样品测序衰减
可能是由于特殊结构如Poly结构、重复序列、回文结构、发卡结构、GCrich、AT富集等导致的测序衰减,由于是样品本身结构问题无法优化建议反向测序进行拼接以得到完整序列,还有一种衰减的情况就是在一段正常峰型后逐渐衰减,可能是模板量反应量不足导致,建议制备高浓度模板测序。
6) 样品测序中断
可能样品存在特殊高级结构,导致dNTP和ddNTP在某一碱基位点后无法与模板结合,测序酶无法继续延伸,建议使用反向引物进行测序经拼接后可以得到完整序列;或酶切后亚克隆测序。
7) 样品测序移码
测序从开端发生移码可能是引物发生降解,建议重新提供引物;测序局部出现移码,可能样品存在特殊高级结构,建议反向测通。
8) 样品测序底峰干扰
可能测序引物不纯,建议将引物进行PAGE胶纯化后在进行测序或重新提供引物测序;可能测序样品不纯,混有正、反向引物,建议重新制备样品测序。
9. 二代测序相关的名词解释
1)高通量测序
高通量测序技术(High-throughputsequencing,HTS)是对传统Sanger测序(称为一代测序技术)革命性的改变, 一次对几十万到几百万条核酸分子进行序列测定, 因此在有些文献中称其为下一代测序技术(next generation sequencing,NGS )足见其划时代的改变, 同时高通量测序使得对一个物种的转录组和基因组进行细致全貌的分析成为可能, 所以又被称为深度测序(Deep sequencing)。
2) 基因组重测序(Genome Re-sequencing)
全基因组重测序是对基因组序列已知的个体进行基因组测序,并在个体或群体水平上进行差异性分析的方法。随着基因组测序成本的不断降低,人类疾病的致病突变研究由外显子区域扩大到全基因组范围。通过构建不同长度的插入片段文库和短序列、双末端测序相结合的策略进行高通量测序,实现在全基因组水平上检测疾病关联的常见、低频、甚至是罕见的突变位点,以及结构变异等,具有重大的科研和产业价值。
3) de novo测序
de novo测序也称为从头测序:其不需要任何现有的序列资料就可以对某个物种进行测序,利用生物信息学分析手段对序列进行拼接,组装,从而获得该物种的基因组图谱。获得一个物种的全基因组序列是加快对此物种了解的重要捷径。随着新一代测序技术的飞速发展,基因组测序所需的成本和时间较传统技术都大大降低,大规模基因组测序渐入佳境,基因组学研究也迎来新的发展契机和革命性突破。利用新一代高通量、高效率测序技术以及强大的生物信息分析能力,可以高效、低成本地测定并分析所有生物的基因组序列。
4) 外显子测序(whole exon sequencing)
外显子组测序是指利用序列捕获技术将全基因组外显子区域DNA捕捉并富集后进行高通量测序的基因组分析方法。外显子测序相对于基因组重测序成本较低,对研究已知基因的SNP、Indel等具有较大的优势,但无法研究基因组结构变异如染色体断裂重组等。
5) mRNA测序(RNA-seq)
转录组学(transcriptomics)是在基因组学后新兴的一门学科,即研究特定细胞在某一功能状态下所能转录出来的所有RNA(包括mRNA和非编码RNA)的类型与拷贝数。Illumina提供的mRNA测序技术可在整个mRNA领域进行各种相关研究和新的发现。mRNA测序不对引物或探针进行设计,可自由提供关于转录的客观和权威信息。研究人员仅需要一次试验即可快速生成完整的poly-A尾的RNA完整序列信息,并分析基因表达、cSNP、全新的转录、全新异构体、剪接位点、等位基因特异性表达和罕见转录等最全面的转录组信息。简单的样品制备和数据分析软件支持在所有物种中的mRNA测序研究。
6) small RNA测序
SmallRNA(micro RNAs、siRNAs和 pi RNAs)是生命活动重要的调控因子,在基因表达调控、生物个体发育、代谢及疾病的发生等生理过程中起着重要的作用。Illumina能够对细胞或者组织中的全部Small RNA进行深度测序及定量分析等研究。实验时首先将18-30 nt范围的Small RNA从总RNA中分离出来,两端分别加上特定接头后体外反转录做成cDNA再做进一步处理后,利用测序仪对DNA片段进行单向末端直接测序。通过Illumina对Small RNA大规模测序分析,可以从中获得物种全基因组水平的miRNA图谱,实现包括新miRNA分子的挖掘,其作用靶基因的预测和鉴定、样品间差异表达分析、miRNAs聚类和表达谱分析等科学应用。
7) miRNA测序
成熟的microRNA(miRNA)是17~24nt的单链非编码RNA分子,通过与mRNA相互作用影响目标mRNA的稳定性及翻译,最终诱导基因沉默,调控着基因表达、细胞生长、发育等生物学过程。基于第二代测序技术的microRNA测序,可以一次性获得数百万条microRNA序列,能够快速鉴定出不同组织、不同发育阶段、不同疾病状态下已知和未知的microRNA及其表达差异,为研究microRNA对细胞进程的作用及其生物学影响提供了有力工具。
8) Chip-seq染色质免疫共沉淀技术(ChromatinImmunoprecipitation,ChIP)也称结合位点分析法,是研究体内蛋白质与DNA相互作用的有力工具,通常用于转录因子结合位点或组蛋白特异性修饰位点的研究。将ChIP与第二代测序技术相结合的ChIP-Seq技术,能够高效地在全基因组范围内检测与组蛋白、转录因子等互作的DNA区段。ChIP-Seq的原理是:首先通过染色质免疫共沉淀技术(ChIP)特异性地富集目的蛋白结合的DNA片段,并对其进行纯化与文库构建;然后对富集得到的DNA片段进行高通量测序。研究人员通过将获得的数百万条序列标签精确定位到基因组上,从而获得全基因组范围内与组蛋白、转录因子等互作的DNA区段信息。
9) CHIRP-SeqCHIRP-Seq( Chromatin Isolationby RNA Purification )是一种检测与RNA绑定的DNA和蛋白的高通量测序方法。方法是通过设计生物素或链霉亲和素探针,把目标RNA拉下来以后,与其共同作用的DNA染色体片段就会附在到磁珠上,最后把染色体片段做高通量测序,这样会得到该RNA能够结合到在基因组的哪些区域,但由于蛋白测序技术不够成熟,无法知道与该RNA结合的蛋白。
10) RIP-seq
RNA Immunoprecipitation是研究细胞内RNA与蛋白结合情况的技术,是了解转录后调控网络动态过程的有力工具,能帮助我们发现miRNA的调节靶点。这种技术运用针对目标蛋白的抗体把相应的RNA-蛋白复合物沉淀下来,然后经过分离纯化就可以对结合在复合物上的RNA进行测序分析。RIP可以看成是普遍使用的染色质免疫沉淀ChIP技术的类似应用,但由于研究对象是RNA-蛋白复合物而不是DNA-蛋白复合物,RIP实验的优化条件与ChIP实验不太相同(如复合物不需要固定,RIP反应体系中的试剂和抗体绝对不能含有RNA酶,抗体需经RIP实验验证等等)。RIP技术下游结合microarray技术被称为RIP-Chip,帮助我们更高通量地了解癌症以及其它疾病整体水平的RNA变化。
11) CLIP-seq
CLIP-seq,又称为HITS-CLIP,即紫外交联免疫沉淀结合高通量测序(crosslinking-immunprecipitationand high-throughput sequencing),是一项在全基因组水平揭示RNA分子与RNA结合蛋白相互作用的革命性技术。其主要原理是基于RNA分子与RNA结合蛋白在紫外照射下发生耦联,以RNA结合蛋白的特异性抗体将RNA-蛋白质复合体沉淀之后,回收其中的RNA片段,经添加接头、RT-PCR等步骤,对这些分子进行高通量测序,再经生物信息学的分析和处理、总结,挖掘出其特定规律,从而深入揭示RNA结合蛋白与RNA分子的调控作用及其对生命的意义。
12) 染色体构象捕获技术
3C 通常是用启动子或者某一个基因或者基因组某一个短的片段在邻近的几十kb或者几百kb基因组扫描可以获得相互作用区域。由于实验需要特异性引物,因而实验室相当费力的,且检测范围小。4C同3C一样做单位点的检测,但其检测扩展到了整个基因组上。主要是引入了反向PCR,因而只需要对这一单一位点设计引物即可。5C 做两个大片段之间相互作用点的检测,可以达到10Mb水平。其仍需使用引物,且引物设计是其技术的难点。Hi-C 可以实现基因组对基因组水平的检测,但是获得高精度需要非常大的测序深度。ChIA-PET标在于特定的蛋白因子及其相关联的染色质相互作用.该技术将配对末端标签测序技术与ChIP相结合, 对富集了某种蛋白质的DNA 片段进行交联, 可以测定全基因组范围的特定转录因子参与的染色质远程交互作用, 从而可以呈现高特异性和高分辨率的染色质相互作用。
13) Hi-C辅助基因组组装
Hi-C辅助基因组组装是指在已有二代或三代或光学图谱辅助组装的Draft genome序列和已知染色体数目的前提下,利用Hi-C测序数据将Draft genome序列进行染色体群组的划分,并确定各序列在染色体上的顺序和方向,使基因组组装组装水平提升到染色体水平的技术。
14) metagenomic(宏基因组)
Magenomics研究的对象是整个微生物群落。相对于传统单个细菌研究来说,它具有众多优势,其中很重要的两点:(1) 微生物通常是以群落方式共生于某一小生境中,它们的很多特性是基于整个群落环境及个体间的相互影响的,因此做Metagenomics研究比做单个个体的研究更能发现其特性;(2)Metagenomics研究无需分离单个细菌,可以研究那些不能被实验室分离培养的微生物。宏基因组是基因组学一个新兴的科学研究方向。宏基因组学(又称元基因组学,环境基因组学,生态基因组学等),是研究直接从环境样本中提取的基因组遗传物质的学科。传统的微生物研究依赖于实验室培养,宏基因组的兴起填补了无法在传统实验室中培养的微生物研究的空白。过去几年中,DNA测序技术的进步以及测序通量和分析方法的改进使得人们得以一窥这一未知的基因组科学领域。
15) SNP、SNV(单核苷酸位点变异)
单核苷酸多态性singlenucleotide polymorphism,SNP 或单核苷酸位点变异SNV。个体间基因组DNA序列同一位置单个核苷酸变异(替代、插入或缺失)所引起的多态性。不同物种、个体基因组DNA序列同一位置上的单个核苷酸存在差别的现象。有这种差别的基因座、DNA序列等可作为基因组作图的标志。人基因组上平均约每1000个核苷酸即可能出现1个单核苷酸多态性的变化,其中有些单核苷酸多态性可能与疾病有关,但可能大多数与疾病无关。单核苷酸多态性是研究人类家族和动植物品系遗传变异的重要依据。在研究癌症基因组变异时,相对于正常组织,癌症中特异的单核苷酸变异是一种体细胞突变(somatic mutation),称做SNV。
16) INDEL (基因组小片段插入)
基因组上小片段(>50bp)的插入或缺失,形同SNP/SNV。
17) copy number variation(CNV):基因组拷贝数变异
基因组拷贝数变异是基因组变异的一种形式,通常使基因组中大片段的DNA形成非正常的拷贝数量。例如人类正常染色体拷贝数是2,有些染色体区域拷贝数变成1或3,这样,该区域发生拷贝数缺失或增加,位于该区域内的基因表达量也会受到影响。如果把一条染色体分成A-B-C-D四个区域,则A-B-C-C-D/A-C-B-C-D/A-C-C-B-C-D/A-B-D分别发生了C区域的扩增及缺失,扩增的位置可以是连续扩增如A-B-C-C-D也可以是在其他位置的扩增,如A-C-B-C-D。
18) structure variation(SV):基因组结构变异
染色体结构变异是指在染色体上发生了大片段的变异。主要包括染色体大片段的插入和缺失(引起CNV的变化),染色体内部的某块区域发生翻转颠换,两条染色体之间发生重组(inter-chromosometrans-location)等。一般SV的展示利用Circos软件。
19) Segment duplication
一般称为SD区域,串联重复是由序列相近的一些DNA片段串联组成。串联重复在人类基因多样性的灵长类基因中发挥重要作用。在人类染色体Y和22号染色体上,有很大的SD序列。
20) genotype and phenotype
既基因型与表型;一般指某些单核苷酸位点变异与表现形式间的关系。
21) Read
高通量测序平台产生的短序列就称为reads。PE125,就是读长为125bp双端测序。
22) Contig
拼接软件基于reads之间的overlap区,拼接获得的序列称为Contig(重叠群),无N。
23) Scaffold
基因组de novo测序,通过reads拼接获得Contigs后,往往还需要构建454 Paired-end库或Illumina Mate-pair库,以获得一定大小片段(如3Kb、6Kb、10Kb、20Kb)两端的序列。基于这些序列,可以确定一些Contig之间的顺序关系,这些先后顺序已知的Contigs组成Scaffold(含有N)。
24) Contig N50
Reads拼接后会获得一些不同长度的Contigs。将所有的Contig长度相加,能获得一个Contig总长度。然后将所有的Contigs按照从长到短进行排序,如获得Contig 1,Contig 2,Contig 3...………Contig 25。将Contig按照这个顺序依次相加,当相加的长度达到Contig总长度的一半时,最后一个加上的Contig长度即为Contig N50。举例:Contig 1+Contig 2+ Contig 3+Contig4=Contig总长度*1/2时,Contig 4的长度即为Contig N50。Contig N50可以作为基因组拼接的结果好坏的一个判断标准。
25) Scaffold N50
Scaffold N50与Contig N50的定义类似。Contigs拼接组装获得一些不同长度的Scaffolds。将所有的Scaffold长度相加,能获得一个Scaffold总长度。然后将所有的Scaffolds按照从长到短进行排序,如获得Scaffold 1,Scaffold 2,Scaffold 3...………Scaffold 25。将Scaffold按照这个顺序依次相加,当相加的长度达到Scaffold总长度的一半时,最后一个加上的Scaffold长度即为Scaffold N50。举例:Scaffold 1+Scaffold 2+Scaffold 3 +Scaffold 4 +Scaffold 5=Scaffold总长度*1/2时,Scaffold 5的长度即为Scaffold N50。Scaffold N50可以作为基因组拼接的结果好坏的一个判断标准。
26) 测序深度和覆盖度
测序深度是指测序得到的总碱基数与待测基因组大小的比值。假设一个基因大小为2M,测序深度为10X,那么获得的总数据量为20M。覆盖度是指测序获得的序列占整个基因组的比例。由于基因组中的高GC、重复序列等复杂结构的存在,测序最终拼接组装获得的序列往往无法覆盖有所的区域,这部分没有获得的区域就称为Gap。例如一个细菌基因组测序,覆盖度是98%,那么还有2%的序列区域是没有通过测序获得的。
27) RPKM、FPKM
RPKM, Reads Per Kilobase of exon model per Million mapped reads每1百万个map上的reads中map到外显子的每1K个碱基上的reads个数。假如有1百万个reads映射到了人的基因组上,那么具体到每个外显子呢,有多少映射上了呢,而外显子的长度不一,那么每1K个碱基上又有多少reads映射上了呢,这大概就是这个RPKM的直观解释。如果对应特定基因的话,那么就是每1000000 mapped到该基因上的reads中每kb有多少是mapped到该基因上的exon的read。
28) Total exon reads
映射到外显子上总的reads个数。这个是映射到某个区域上的reads个数,这个区域或者是已知注释的基因或者跨两个外显子的边界或者是某个基因已经注释的转录本的内含子、外显子。对于真核生物来说,外显子和它们自己内部的关系由某类型的mRNA来注释。
29) Exon length:外显子的长度
计算时,计算所有某个基因已注释的所有外显子长度的总和。即使某个基因以多种注释的转录本呈现,这个外显子在求和时只被包含一次。即使部分重叠的外显子共享相同的区域,重叠的外显子以其总长来计算。
30) Mapped reads
映射到某个基因上的所有reads总数。因此这包含所有的唯一映射到这个区域上的reads。举例:比如对应到该基因的read有1000个,总reads个数有100万,而该基因的外显子总长为5kb,那么它的RPKM为:10^9*1000(reads个数)/10^6(总reads个数)*5000(外显子长度)=200或者:1000(reads个数)/1(百万)*5(K)=200这个值反映基因的表达水平。
31) FPKM (fragmentsper kilobase of exon per million fragments mapped)
FPKM与RPKM计算方法基本一致。不同点就是FPKM计算的是fragments,而RPKM计算的是reads。Fragment比read的含义更广,因此FPKM包含的意义也更广,可以是pair-end的一个fragment,也可以是一个read。
32) 转录本重构
用测序的数据组装成转录本。有两种组装方式:1,de-novo构建; 2,有参考基因组重构。其中de-novo组装是指在不依赖参考基因组的情况下,将有overlap的reads连接成一个更长的序列,经过不断的延伸,拼成一个个的contig及scaffold。常用工具包括velvet,trans-ABYSS,Trinity等。有参考基因组重构,是指先将read贴回到基因组上,然后在基因组通过reads覆盖度,junction位点的信息等得到转录本,常用工具包括scripture、cufflinks。
33) 表达谱
基因表达谱(geneexpression profile):指通过构建处于某一特定状态下的细胞或组织的非偏性cDNA文库,大规模cDNA测序,收集cDNA序列片段、定性、定量分析其mRNA群体组成,从而描绘该特定细胞或组织在特定状态下的基因表达种类和丰度信息,这样编制成的数据表就称为基因表达谱
34) 比较基因组学
比较基因组学(ComparativeGenomics)是基于基因组图谱和测序基础上,对已知的基因和基因组结构进行比较,来了解基因的功能、表达机理和物种进化的学科。利用模式生物基因组与人类基因组之间编码顺序上和结构上的同源性,克隆人类疾病基因,揭示基因功能和疾病分子机制,阐明物种进化关系,及基因组的内在结构。
35) 基因组注释
基因组注释(Genomeannotation)是利用生物信息学方法和工具,对基因组所有基因的生物学功能进行高通量注释,是当前功能基因组学研究的一个热点。基因组注释的研究内容包括基因识别和基因功能注释两个方面。基因识别的核心是确定全基因组序列中所有基因的确切位置。
10. 三代全长转录本分析工具
三代全长转录本在辅助基因注释,可变剪接分析,融合基因检测方面可以说大显身手,以下几个工具及对应的下载地址。
1) 可变剪接鉴定(3个工具)
(1)网址:https://github.com/liuxiaoxian/IsoSeq_AS_de_novo
Liu X, Mei W, Soltis P S, et al. Detecting Alternatively Spliced Transcript Isoforms from Single‐Molecule Long‐Read Sequences without a Reference Genome[J]. Molecular Ecology Resources, 2017.
(2)网址:http://splicegrapher.sourceforge.net/
Rogers M F, Thomas J, Reddy A S N, et al. SpliceGrapher: detecting patterns of alternative splicing from RNA-Seq data in the context of gene models and EST data[J]. Genome biology, 2012, 13(1): R4.
(3)网址:https://sourceforge.net/projects/cash-program/
Wu W, Zong J, Wei N, et al. CASH: a constructing comprehensive splice site method for detecting alternative splicing events[J]. Briefings in Bioinformatics, 2017: bbx034.
2) 多平台结合分析高基因密度基因组
网址:https://github.com/flemingtonlab/public
O’Grady T, Wang X, Höner Zu Bentrup K, Baddoo M, Concha M, Flemington EK. Global transcript structure resolution of high gene density genomes through multi-platform data integration. Nucleic Acids Res. 2016 Jul 12; PMID: 27407110.
3) 全长转录本分析流程TAPIS
网址:https://bitbucket.org/comp_bio/tapis
Abdel-Ghany S E, Hamilton M, Jacobi J L, et al. A survey of the sorghum transcriptome using single-molecule long reads[J]. Nature communications, 2016, 7.
4) 全长转录组浏览器
网址:https://github.com/goeckslab/isoseq-browser
Hu J, Uapinyoying P, Goecks J. Interactive analysis of Long-read RNA isoforms with Iso-Seq Browser[J]. bioRxiv, 2017: 102905.
5) 全长转录组测序新转录结构发现注释工具
网址:https://bitbucket.org/ConesaLab/sqanti
Tardaguila M, de la Fuente L, Marti C, et al. SQANTI: extensive characterization of long read transcript sequences for quality control in full-length transcriptome identification and quantification[J]. bioRxiv, 2017: 118083.
6) 全长转录组Iso-Seq和RNA-Seq集合进行无参考转录组分析
Ning G, Cheng X, Luo P, et al. Hybrid sequencing and map finding (HySeMaFi): optional strategies for extensively deciphering gene splicing and expression in organisms without reference genome[J]. Scientific Reports, 2017, 7.
参考文献
1. https://doi.org/10.1007/s40142-021-00199-x
2.《高通量测序技术》
3. Nurk, Sergey, et al. The complete sequence of a human genome. bioRxiv (2021).
4. Logsdon, G.A., Vollger, M.R. & Eichler, E.E. Long-read human genome sequencing and its applications. Nat Rev Genet 21, 597–614 (2020)
5. Midha, M. K. et al. Long-read sequencing in deciphering human genetics to a greater depth. Human Genetics(2019).
6. Roberts RJ, Carneiro MO, Schatz MC. The advantages of SMRT sequencing [published correction appears in Genome Biol. 2017 Aug 16;18(1):156]. Genome Biol. 2013;14(7):405. Published 2013 Jul 3. doi:10.1186/gb-2013-14-6-405.
7. Mikheyev AS, Tin MM. A first look at the Oxford Nanopore MinION sequencer. Mol Ecol Resour. 2014;14(6):1097-1102. doi:10.1111/1755-0998.12324
8. Metzker ML. Sequencing technologies - the next generation. Nat Rev Genet. 2010;11(1):31-46. doi:10.1038/nrg2626.
9. Shendure J, Ji H. Next-generation DNA sequencing. Nat Biotechnol. 2008;26(10):1135-1145. doi:10.1038/nbt1486.
10. Lin B, Hui J, Mao H. Nanopore Technology and Its Applications in Gene Sequencing. Biosensors (Basel). 2021;11(7):214. Published 2021 Jun 30. doi:10.3390/bios11070214.
11. Sanger, F. & Nicklen, S. DNA sequencing with chain-terminating. 74, 5463–5467 (1977).
12. The Oxford Nanopore MinION: delivery of nanopore sequencing to the genomics community
原文地址:https://zhuanlan.zhihu.com/p/719714888 |
|

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 






 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 发表于 2025-2-18 20:04
发表于 2025-2-18 20:04



