【爆肝万字】基于Rosetta的配体结合位点的设计(Rosetta and the Design of Ligand Binding Sites )
1. 前言
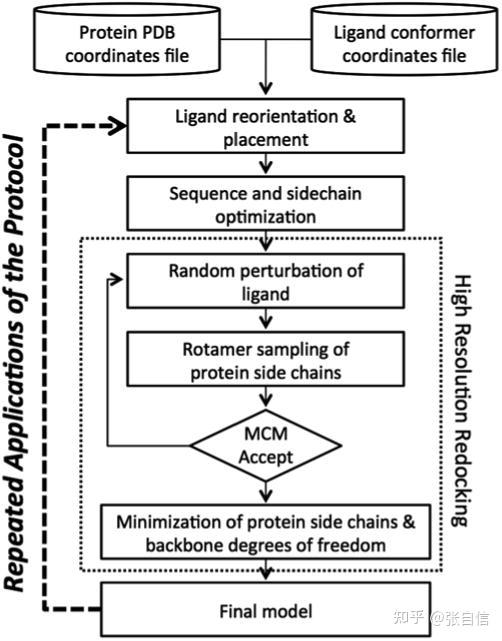
Rosetta是蛋白质设计中的重要平台,在蛋白质设计领域中占有重要地位。本文以2016年的文献Rosetta and the Design of Ligand Binding Sites进行实践,探索用Rosetta进行配体结合位点的设计。经过三个月实践成功,得到本文档,在此分享给读者,希望能够帮到有需要的读者完成实验工作,早发文章! 觉得有用还请点个赞呀!ヾ(◍°∇°◍)ノ゙
文献信息:Methods Mol Biol. 2016 ; 1414: 47–62. doi:10.1007/978-1-4939-3569-7_4. Rosetta and the Design of Ligand Binding Sites - PubMed

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 






 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 发表于 2025-1-22 17:02
发表于 2025-1-22 17:02










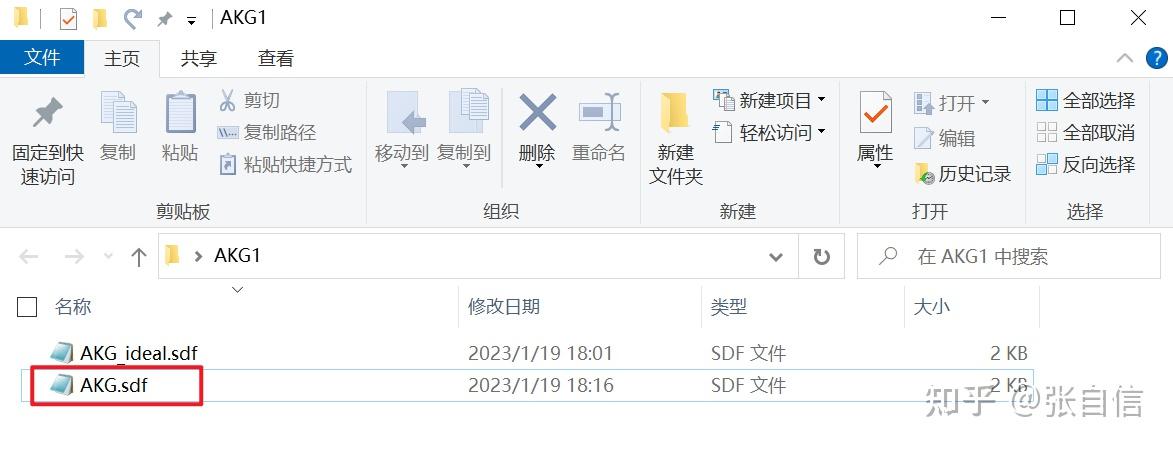
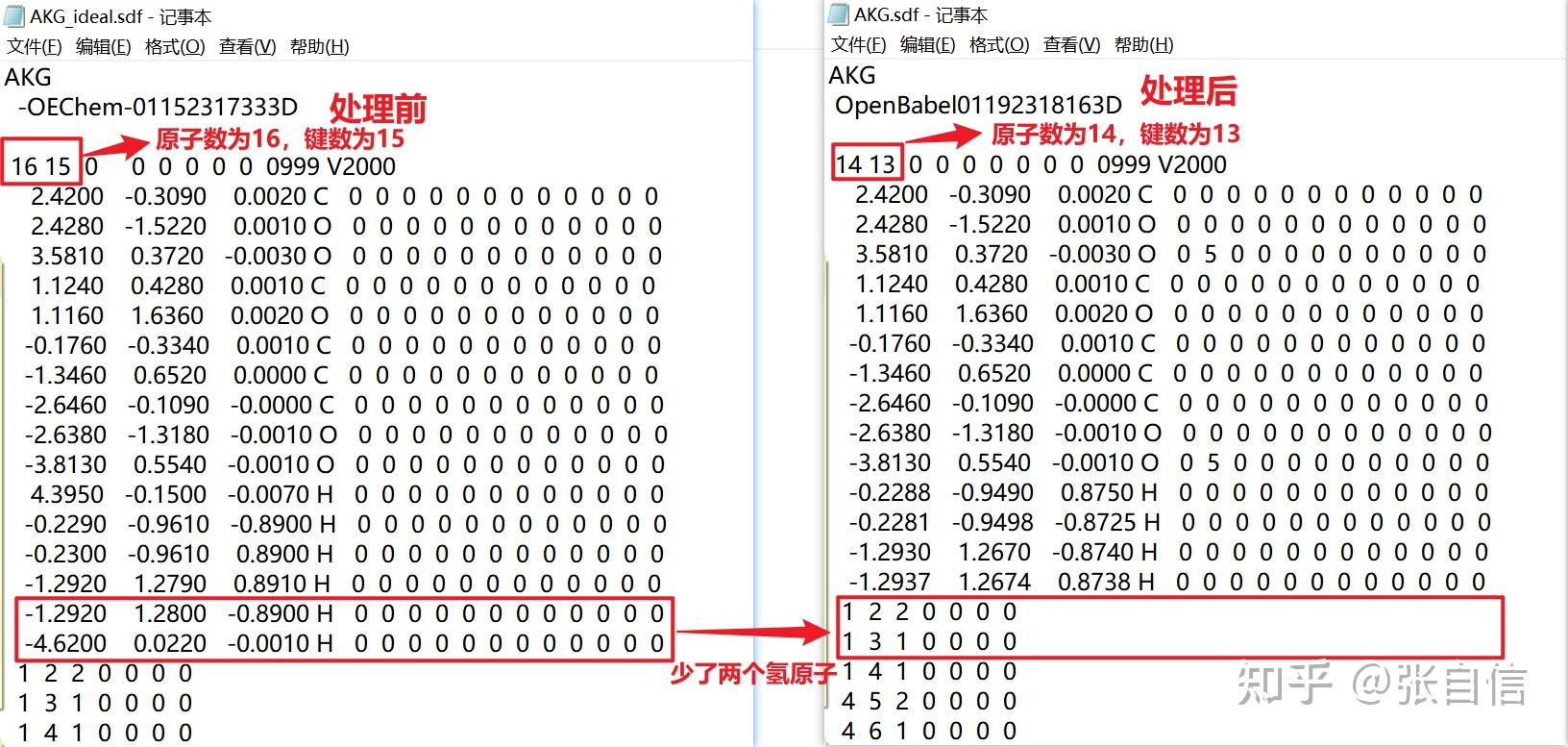
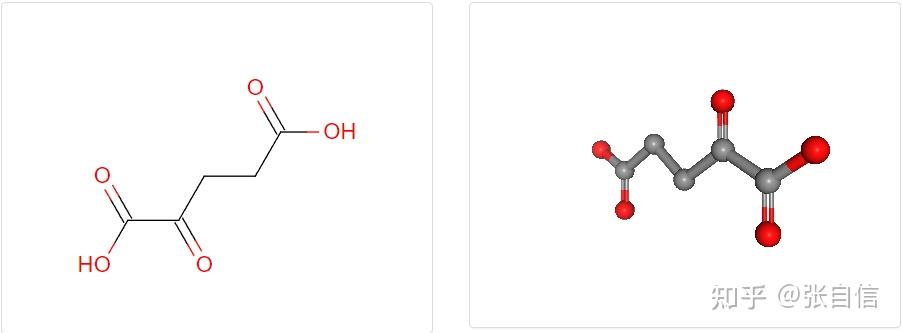

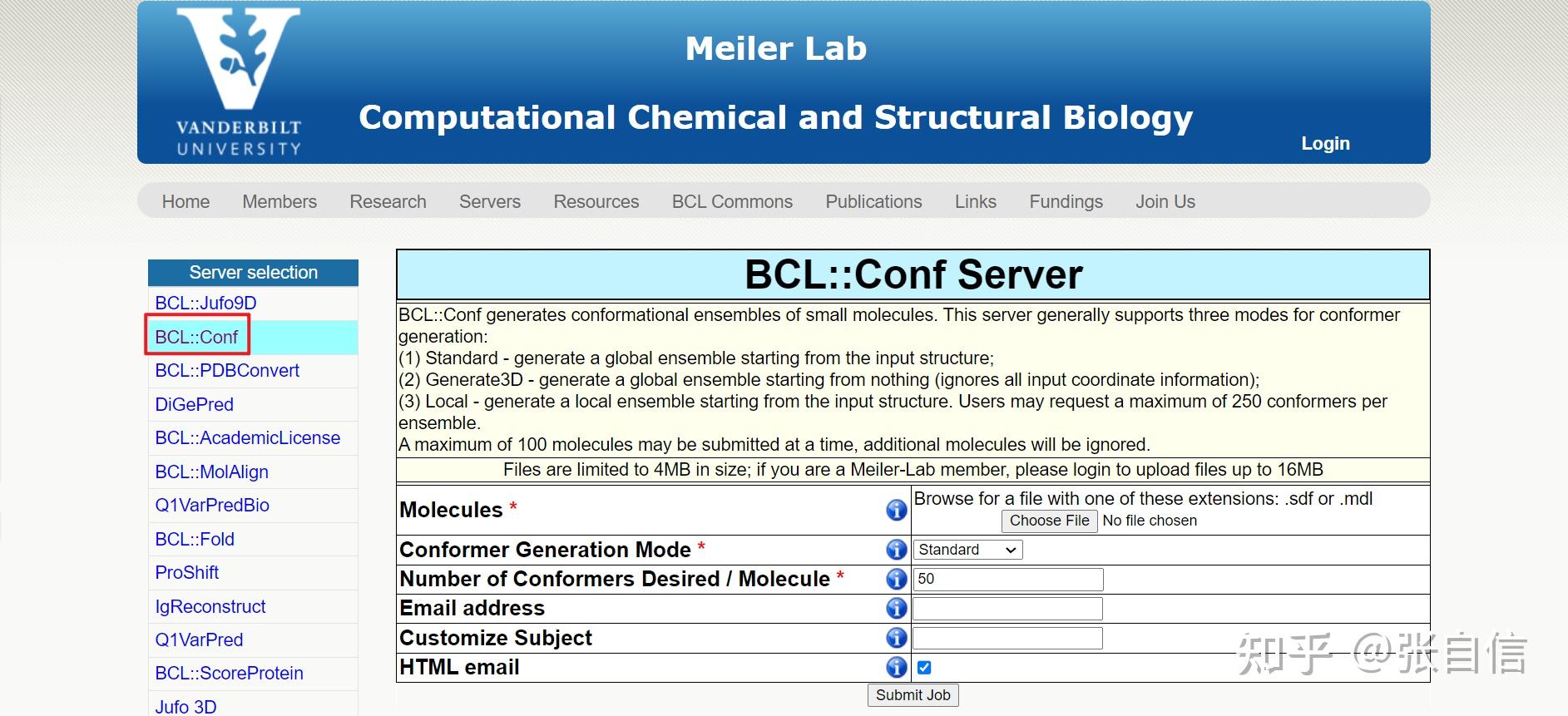
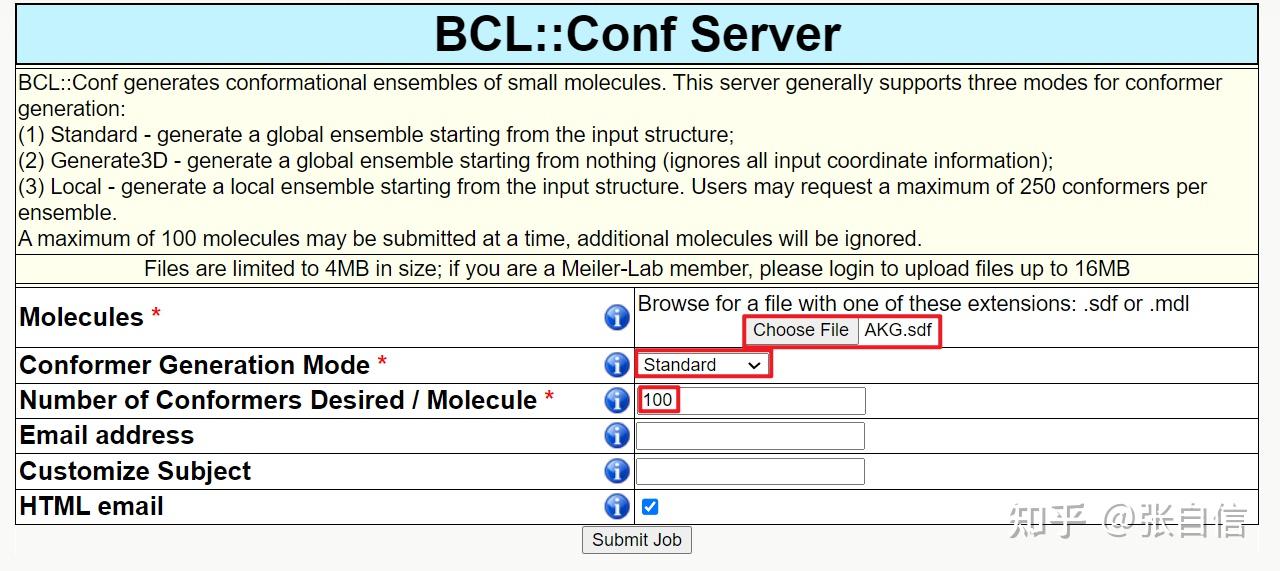
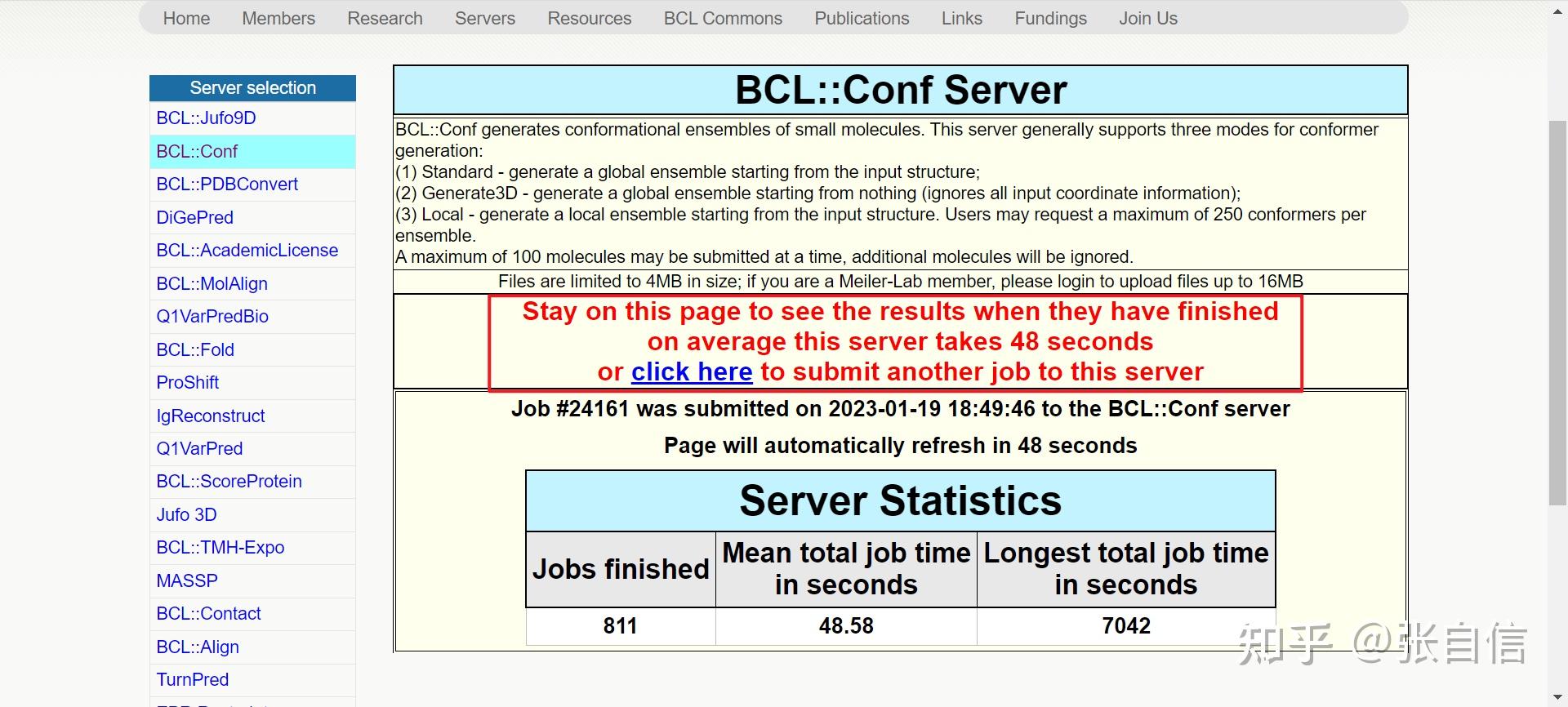
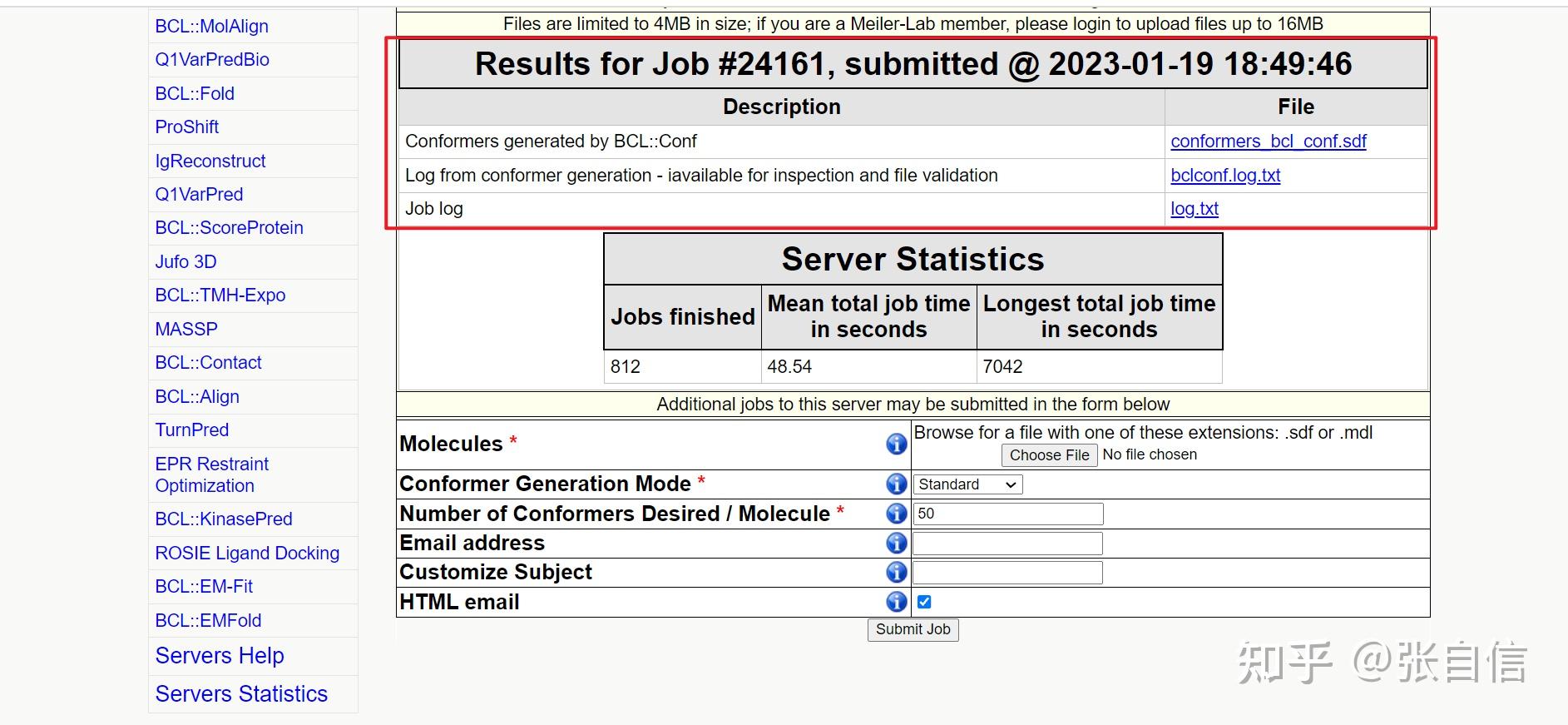
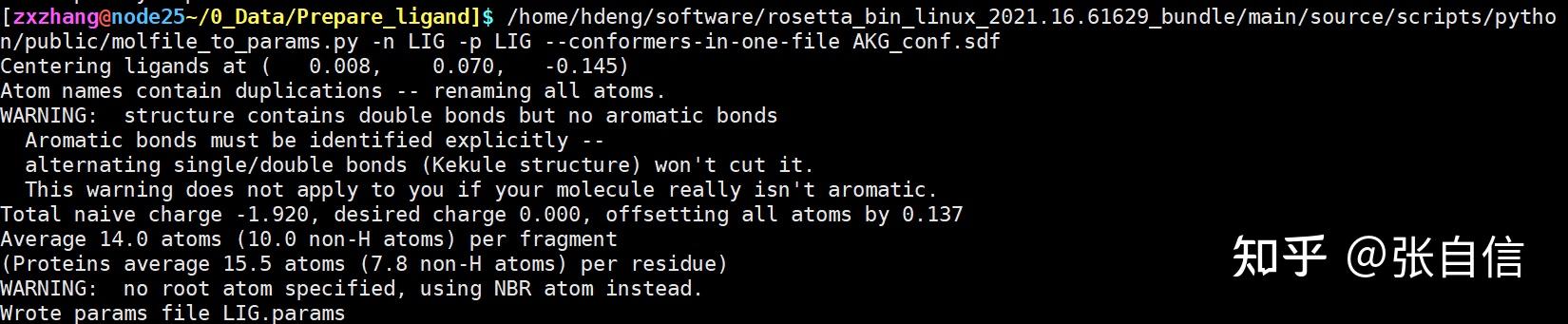
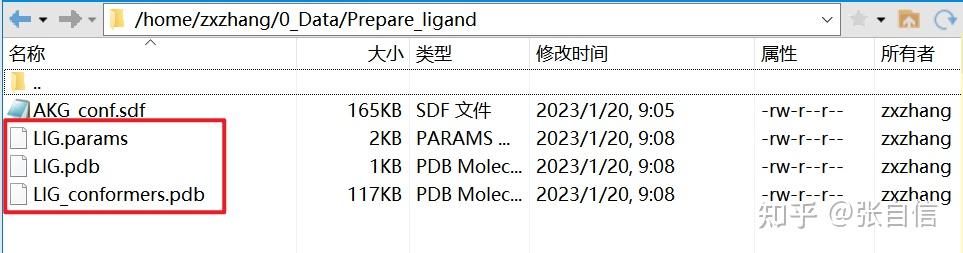
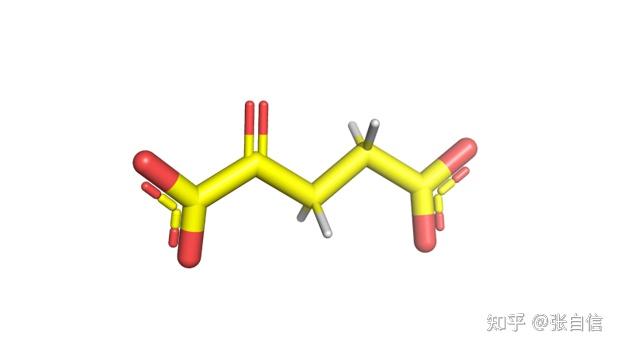

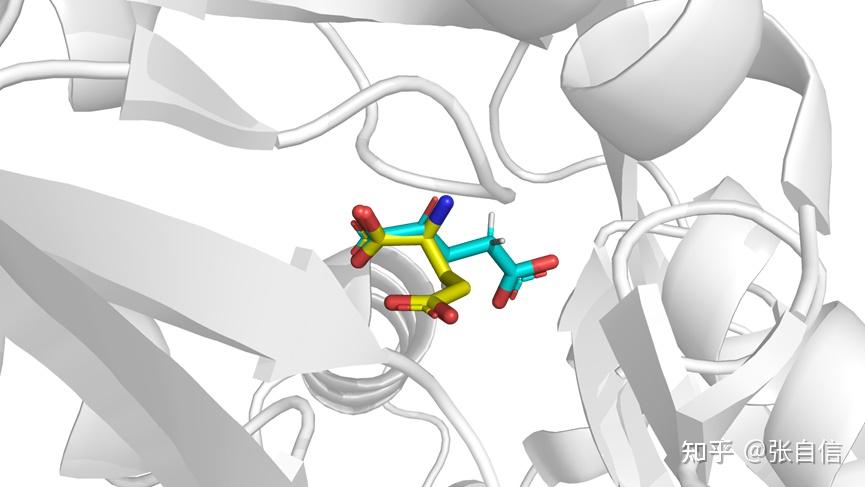
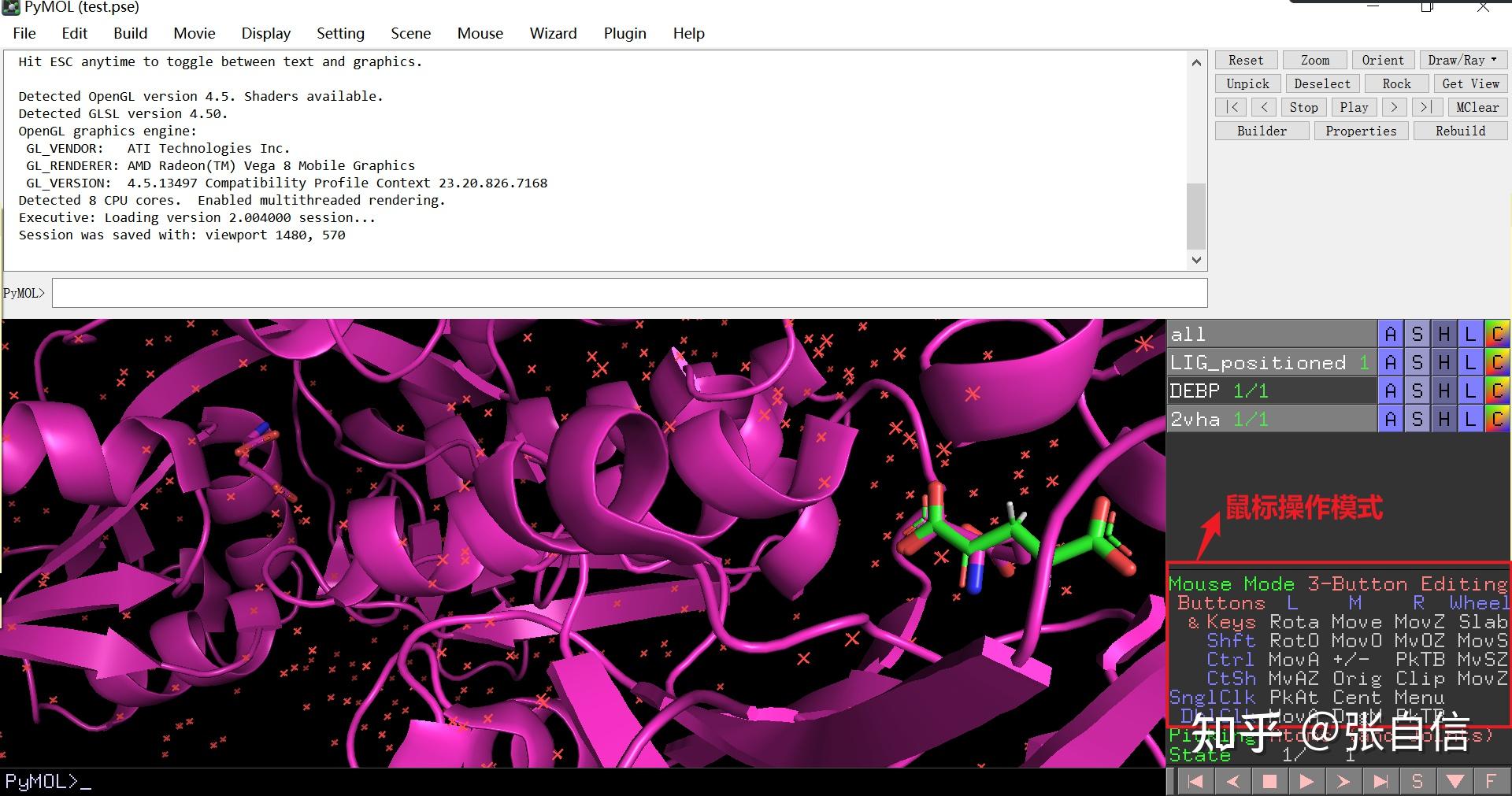
 发表于 2025-1-22 17:05
发表于 2025-1-22 17:05