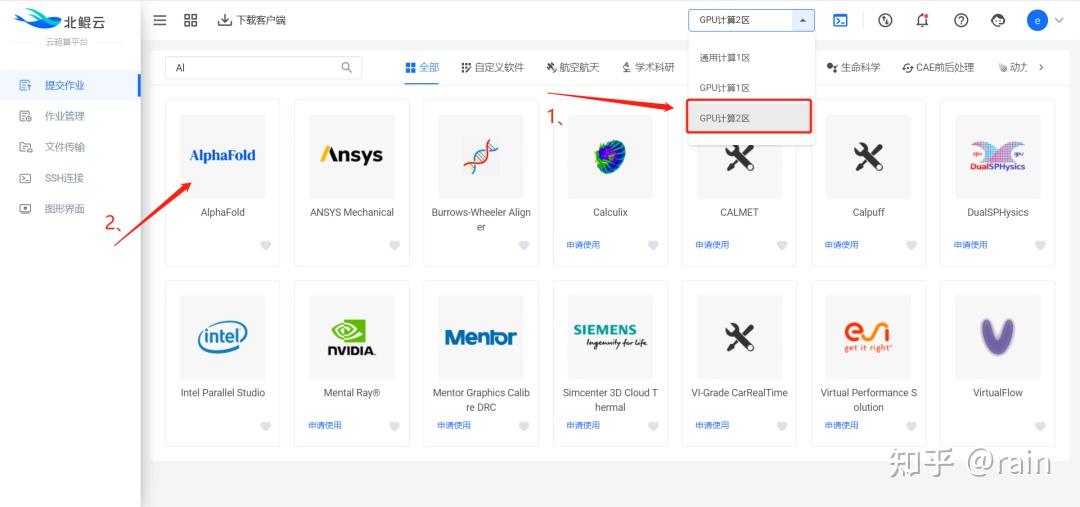
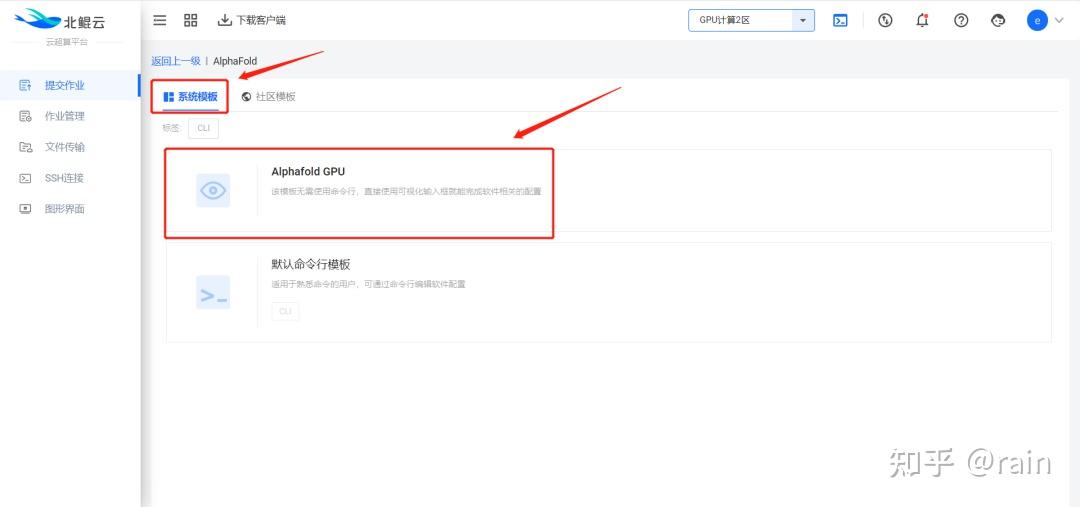
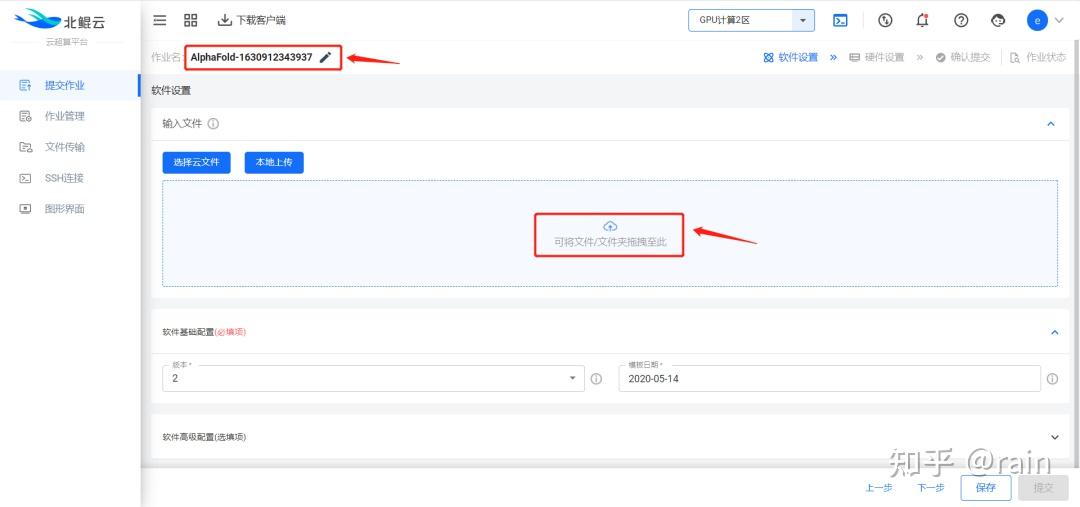
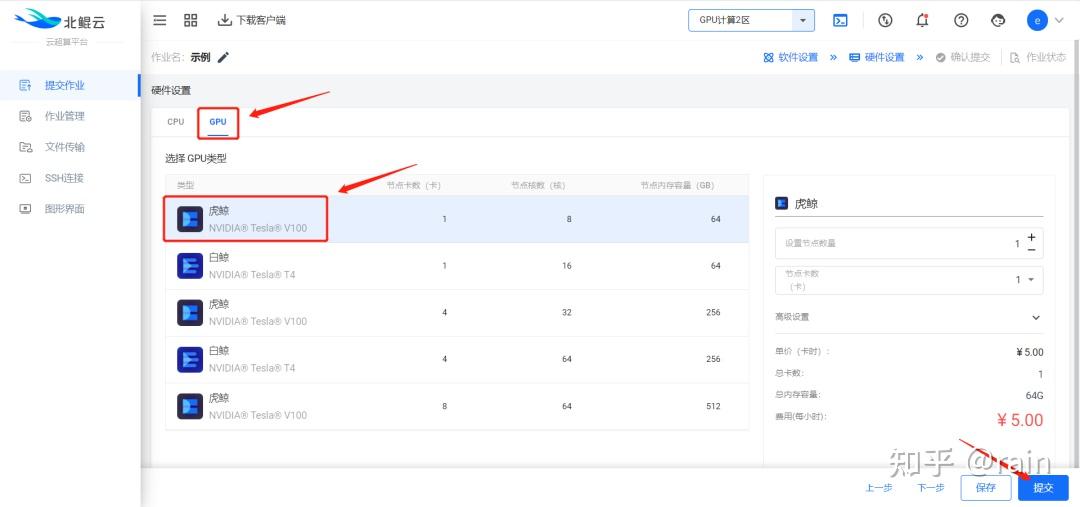
金桔
金币
威望
贡献
回帖0
精华
在线时间 小时
|
蛋白质结构预测方法的目标是利用计算模型来确定蛋白质分子中每个原子的空间位置,仅从其氨基酸序列开始。根据是否可以在蛋白质数据库 (PDB) 中找到同源结构,结构预测方法历来被归类为基于模板的建模 (TBM) 或无模板建模 (FM) 方法。直到最近,TBM 一直是预测蛋白质结构最可靠的方法,并且在缺乏可靠模板的情况下,建模精度急剧下降。尽管如此,最近的蛋白质结构预测实验评估 (CASP14) 的结果表明,蛋白质结构预测问题可以通过使用端到端深度机器学习技术在很大程度上得到解决,其中可以构建正确的折叠适用于几乎所有单域蛋白质,而无需使用 PDB 模板。至关重要的是,模型质量与可用模板结构的质量以及针对给定目标蛋白质检测到的序列同源物的数量几乎没有相关性。因此,深度学习技术的实施基本上突破了 TBM 和 FM 方法之间已有 50 年历史的建模边界,并使高分辨率结构预测的成功大大减少了对 PDB 库中模板可用性的依赖。
使用深度学习的端到端结构预测
端到端折叠的首次尝试
虽然迄今为止许多成功的方法都专注于预测成对结构特征并将其纳入结构组装模拟,但解决结构预测问题的理想方法是从蛋白质的氨基酸序列开始直接学习蛋白质的 3D 结构,因此-称为“端到端”学习方法。这将消除对高级折叠模拟的需要,而是允许深度神经网络直接生成 3D 结构。基于端到端深度学习的结构预测的首批尝试之一是使用循环几何网络通过预测每个残基的骨架扭转角来构建蛋白质模型。在这里,重要的是要注意蛋白质结构可以根据构成每个氨基酸残基的所有原子的笛卡尔坐标或在扭转角空间中描述,假设理想的键长和键角。通过其扭转角表示蛋白质构象允许预测和优化显着更少的参数。此外,使用机器学习更难以预测笛卡尔坐标,因为旋转或平移结构会导致相同蛋白质结构的坐标显着不同。因此,需要一种不依赖于任意平移或旋转的表示来实现自洽,这就是该方法使用蛋白质结构的扭转角表示的原因。然而,扭转角表示的一个缺点是,局部残基的任何小错误都可能导致全局结构的大 RMSD 错误。深度神经网络由堆叠的长短期记忆 (LSTM) 单元组成,这些单元接收位置特定的氨基酸和 PSSM 信息以及来自其他上游和下游 LSTM 单元的信息。网络的输出是每个残基的预测骨架扭转角。根据这些预测的扭转角,通过使用简单的几何函数从扭转角空间转换为笛卡尔空间,可以从 N 端到 C 端一次一个残基直接构建骨架结构。深度神经网络由堆叠的长短期记忆 (LSTM) 单元组成,这些单元接收位置特定的氨基酸和 PSSM 信息以及来自其他上游和下游 LSTM 单元的信息。网络的输出是每个残基的预测骨架扭转角。根据这些预测的扭转角,通过使用简单的几何函数从扭转角空间转换为笛卡尔空间,可以从 N 端到 C 端一次一个残基直接构建骨架结构。深度神经网络由堆叠的长短期记忆 (LSTM) 单元组成,这些单元接收位置特定的氨基酸和 PSSM 信息以及来自其他上游和下游 LSTM 单元的信息。网络的输出是每个残基的预测骨架扭转角。根据这些预测的扭转角,通过使用简单的几何函数从扭转角空间转换为笛卡尔空间,可以从 N 端到 C 端一次一个残基直接构建骨架结构。
虽然这可能是端到端学习的第一个主张之一,但这个想法类似于预测给定查询序列的主干扭转角,这是该领域的一个长期存在的想法。除了神经网络架构之外,主要区别之一是训练的损失函数考虑了预测结构和原生结构之间的偏差,而不仅仅是扭转角预测中的误差。然而,该方法在 CASP13 中表现不佳,表明仅直接预测扭转角可能不是构建三级结构模型的可靠方法。这部分是因为扭转角本质上是局部特征,可能无法准确捕获对结构建模至关重要的远程信息,并且由于杠杆臂效应,预测的扭转角中的小错误可能导致下游的较大结构偏差。实际上。最终,他们使用网络生成短结构片段,然后使用距离图引导的片段组装方法进行组装。
与循环几何网络方法大约同时开发的另一种端到端折叠方法是 NEMO。NEMO 结合使用一维、二维和图形卷积来预测残基间距离、方向和二面角,并利用朗之万动力学基于这些预测特征生成模型。因此,该方法通过骨架二面角以及残基间距离和方向的组合来表示蛋白质构象。这里需要注意的是,与扭转角表示类似,蛋白质结构可以通过完整的成对距离图以独立于 3D 空间中的平移或旋转的方式进行描述,镜像结构除外。尽管采用了独特的方法,但该方法的性能优于使用基于深度学习的约束的更传统的蛋白质折叠方法。。
深度学习对结构建模精度的影响
CASP 实验提供了一种客观的方法,可以用不同类别的蛋白质结构预测技术对三级结构建模和接触预测进行基准测试。CASP7 开始,根据每个目标的 PDB 模板的可用性和质量,在 CASP 期间建模的蛋白质已被分类为 TBM、TBM-easy、TBM-hard、FM/TBM 或 FM,其中 TBM-easy 目标易于识别,高质量模板和 FM 目标通常在 PDB 中缺乏同源模板。在以下部分中的分析,TBM、TBM-easy、TBM-hard、FM/TBM 目标均视为 TBM 目标,FM 目标单独处理。在 CASP 中,预测是由服务器组和人类组产生的。服务器组必须部署完全自动化的管道并在 72 小时内提交他们的结果,同时对其他组的预测完全不知情。另一方面,对于大多数建模目标,人类组有两周的时间,以允许更多的人为干预,例如从服务器组的最终提交中获取见解。由于提供了更长的计算时间以及对所有服务器组的结果的充分了解,人类组通常比使用类似算法的服务器组表现更好。在 服务器组必须部署完全自动化的管道并在 72 小时内提交他们的结果,同时对其他组的预测完全不知情。另一方面,对于大多数建模目标,人类组有两周的时间,以允许更多的人为干预,例如从服务器组的最终提交中获取见解。由于提供了更长的计算时间以及对所有服务器组的结果的充分了解,人类组通常比使用类似算法的服务器组表现更好。在 服务器组必须部署完全自动化的管道并在 72 小时内提交他们的结果,同时对其他组的预测完全不知情。另一方面,对于大多数建模目标,人类组有两周的时间,以允许更多的人为干预,例如从服务器组的最终提交中获取见解。由于提供了更长的计算时间以及对所有服务器组的结果的充分了解,人类组通常比使用类似算法的服务器组表现更好。在 由于提供了更长的计算时间以及对所有服务器组的结果的充分了解,人类组通常比使用类似算法的服务器组表现更好。在 由于提供了更长的计算时间以及对所有服务器组的结果的充分了解,人类组通常比使用类似算法的服务器组表现更好。
使用深度学习提高接触预测精度
最近 CASP 实验中最显着的发展是深度学习策略的使用,特别是将来自深度学习的接触图和距离图结合到结构预测程序中。CASP没有引入距离图预测的类别,但确实有接触图预测比赛。此外,接触图可以通过将距离图折叠成两个 bin 来从距离图中导出。
从 2014 年到 2018 年,由于利用从协同进化数据开始的深度残差神经网络的接触预测器的发展,接触预测精度几乎增加了两倍。
而在CASP14最佳预测的平均精度为75.1%,这类似于最好CASP13预测器,从CASP13显著下降到CASP14。这是关键的进步,因为大多数基于深度学习的接触/距离图预测方法利用共同进化特征,这需要具有许多序列的 MSA 才能可靠地确定每个位置之间的共同进化耦合。
使用深度学习改进三级结构建模
传统上,预测蛋白质结构最可靠的方法是使用 TBM 方法,该方法依赖于从 PDB 库中识别同源模板以对目标序列进行建模。因此,这种 TBM 方法的准确性高度依赖于从 PDB 库中识别高质量模板的能力,当只能识别低质量模板时,建模精度会急剧下降。理论上,FM 方法不受 PDB 库中模板可用性的限制,但它们传统上优于 TBM 方法,特别是对于 PDB 中具有易于识别模板的目标。尽管如此,结合深度学习和最近的端到端学习预测的成对约束,
图1B显示了之前的 CASP 实验以及最近的 FM 和 TBM 目标实验的结果,每个目标的最佳首次提交模型的平均 TM 分数。在这里,TM-score 是一个与序列长度无关的度量,范围为 [0, 1],其中分数 >0.5 表示预测结构和原生结构共享相同的全局拓扑,分数 >0.914 可用作低到中等分辨率的实验精度。从图中可以看出,随着该领域的进步,FM 和 TBM 目标在建模精度上的差距已经缩小。特别是,FM 模型质量的提高可能归因于使用深度学习约束和端到端学习,因为在缺乏合适的模板结构的情况下,可以使用深度学习来指导结构组装模拟。在 CASP7 中,FM 和 TBM 目标的平均 TM 分数分别为 0.38 和 0.80,这导致 TM 分数差距为 0.42。CASP11 发现差距略微缩小至 0.35,FM 和 TBM 目标的平均 TM 分数分别等于 0.47 和 0.82。
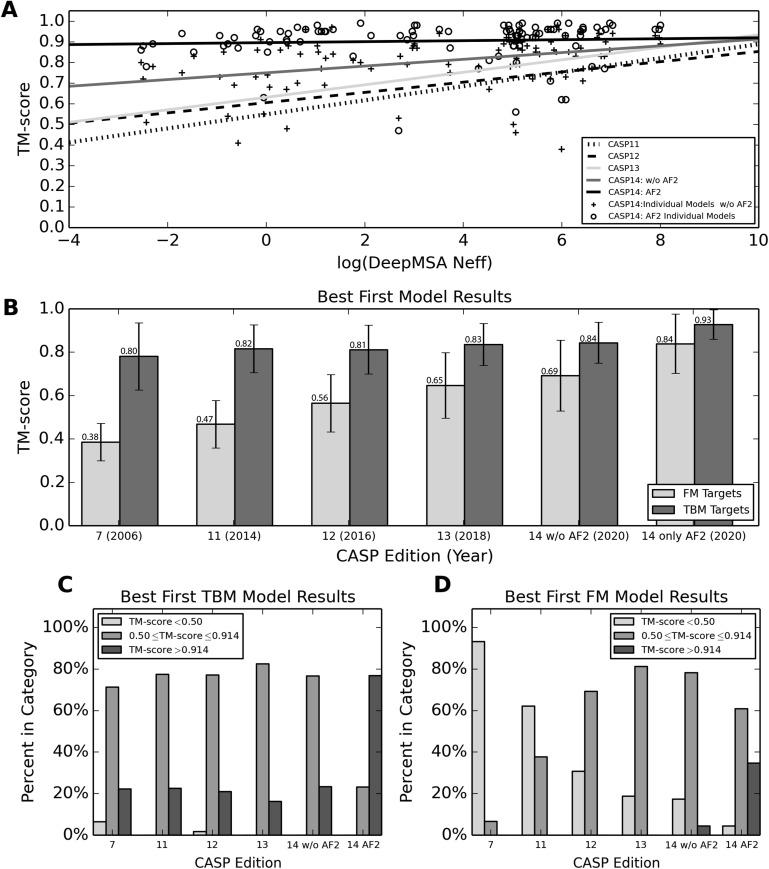
图1
最近 CASP 实验中结构预测结果的总结。(A) 第一个提交模型的最佳 TM 分数与 DeepMSA 程序生成的 MSA 的 Neff 值之间的关系。(B) 在相应的 CASP 比赛中提交的最佳第一个 TBM 和 FM 模型的平均 TM 分数。(C) CASP7/11-14 中任何组提交的最佳首个 TBM 模型(包括 TBM、TBM-easy、TBMA-hard 和 FM/TBM)的结果,其中模型被分为三个类别之一他们的 TM 分数:[0, 0.5), [0.5, 0.914], (0.914, 1.0]. (D) CASP7/11-14 中任何组提交的最佳第一个 FM 模型的结果,其中模型被分类为基于他们的 TM 分数的三个类别之一:[0, 0.5), [0.5, 0.914], (0.914, 1.0]。
深度学习不仅在很大程度上缩小了 TBM 和 FM 方法精度之间的差距,而且还极大地提高了对同源序列很少的目标的建模精度。在 CASP14 中,AlphaFold2 的最终模型质量几乎完全独立于 MSA Neff 值,这是一个真正了不起的成就)。从图1C,还可以看到以实验精度生成的模型数量显着增加(考虑到 0.914 的截止 TM 分数时)。在之前的 CASP 实验中,没有任何 FM 目标能够以如此高的精度折叠,但在 CASP14 中,AlphaFold2 能够以实验精度折叠超过 1/3 的 FM 目标,以及几乎 80% 的 TBM 目标。
需要注意的是,尽管 TBM 和 FM 精度之间的差距已经大大缩小,并且大多数结构预测研究都集中在远同源建模上,其中必须排除近同源模板以促进基准测试和与其他方法的比较,传统的 TBM/FM 和现代深度学习方法本质上依赖于实验求解的结构,因此受到 PDB 中结构数量增加的影响。首先,新解决的实验结构可以为更多序列提供紧密同源的模板,以促进高分辨率 TBM 结构建模。其次,更大的 PDB 结构集包含更全面的折叠类型,这有助于为 FM 开发更强大的基于知识的统计力场和机器学习模型。
参考资料:Pearce R, Zhang Y. Toward the solution of the protein structure prediction problem[J]. Journal of Biological Chemistry, 2021: 100870. |
|

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 






 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 发表于 2025-1-10 11:22
发表于 2025-1-10 11:22


 发表于 2025-1-10 11:22
发表于 2025-1-10 11:22