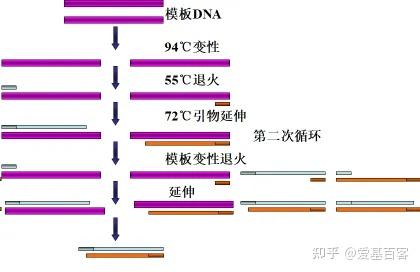
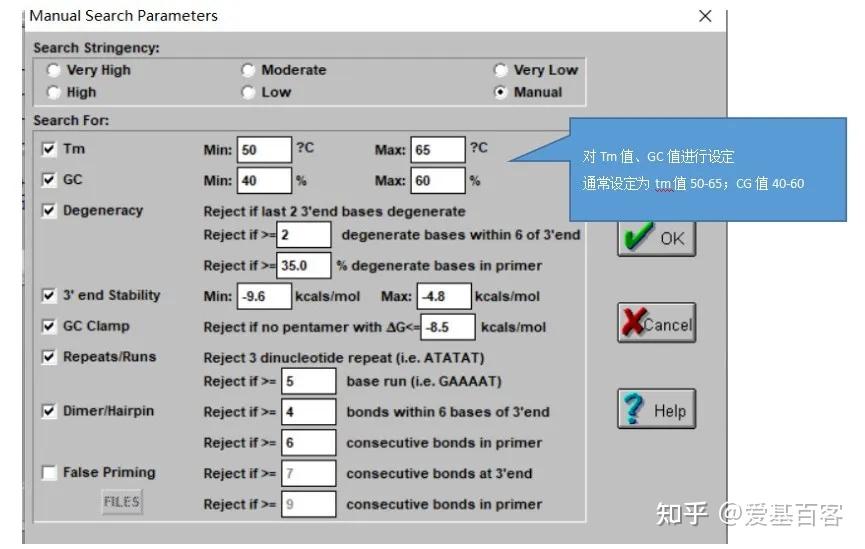
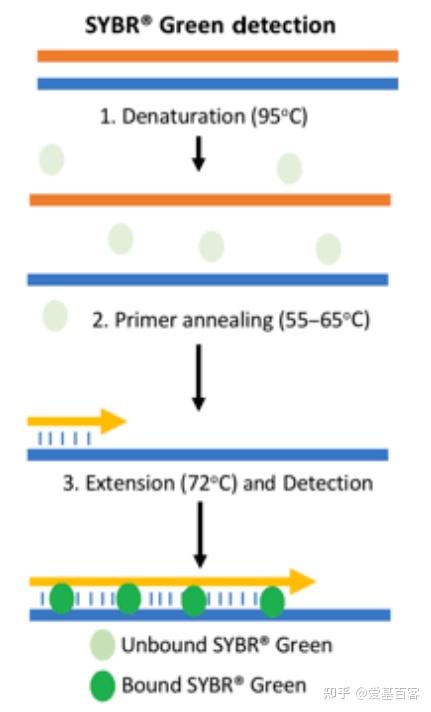
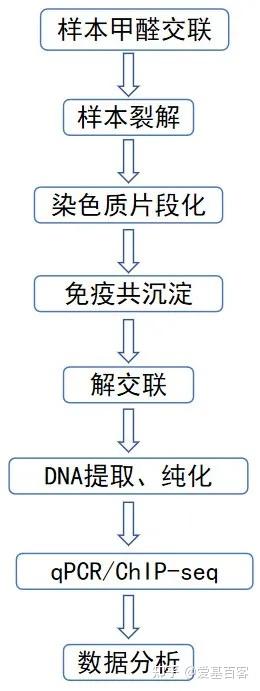
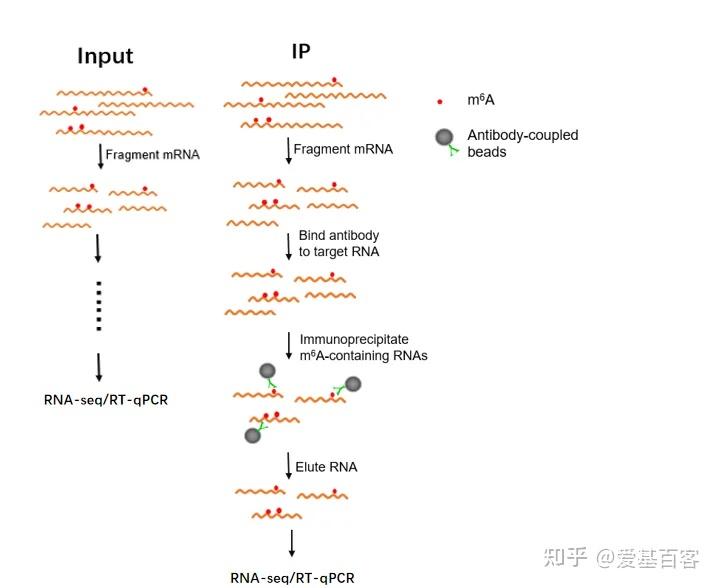
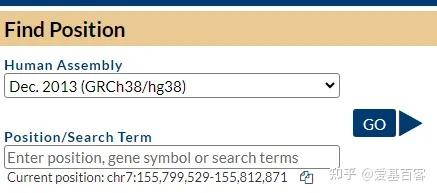
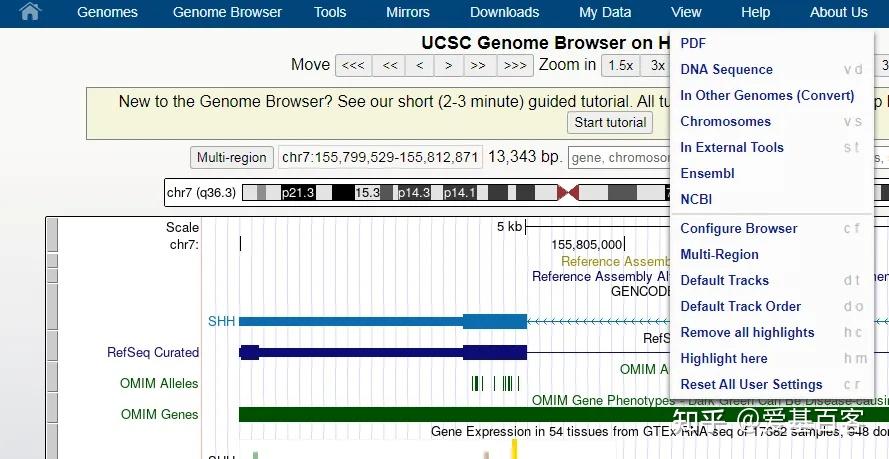
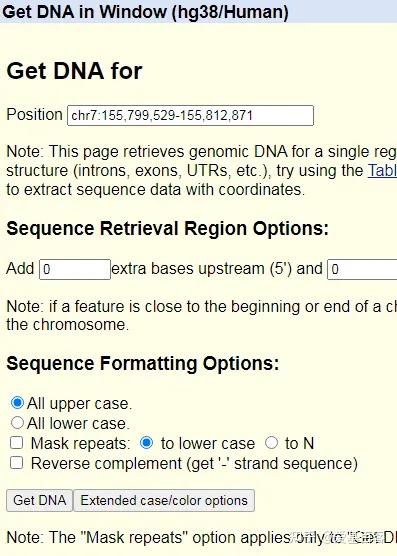
ChIP-qPCR的实验目的不仅包含检测 ChIP 实验是否成功,为后续的建库和测序做准备,还包括检测蛋白与目标区域 DNA 的结合。一般情况下,可以根据已发表文献的ChIP-seq数据,来获取目标区域的引物信息,但当文献中查不到所需要的引物时,我们可以根据目标蛋白结合的靶基因的区域进行设计,比如转录因子通常在启动子区域结合,我们可以在靶基因的启动子区进行引物设计。通常认为基因转录起始位点上游2kb属于基因的启动子区。不过为了保险起见,也可以在多个位置设计引物进行检测。
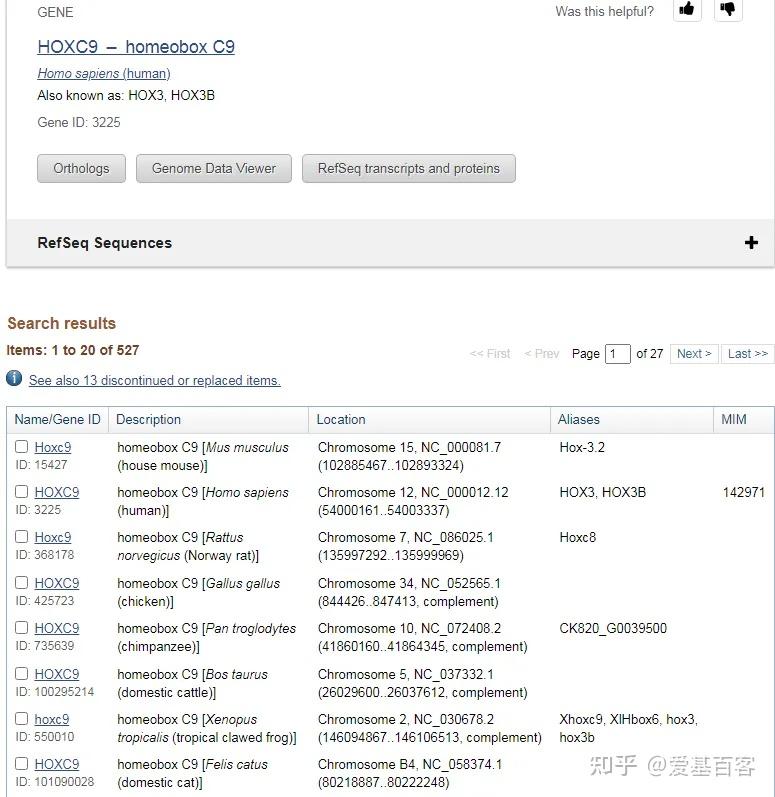
以Hoxc9基因为例,该基因启动子区域碱基序列查询如下:
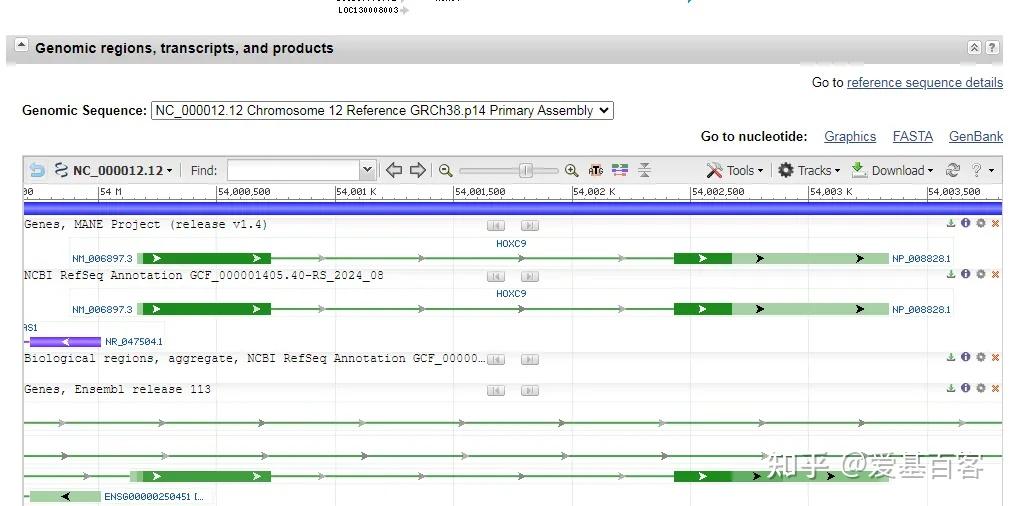
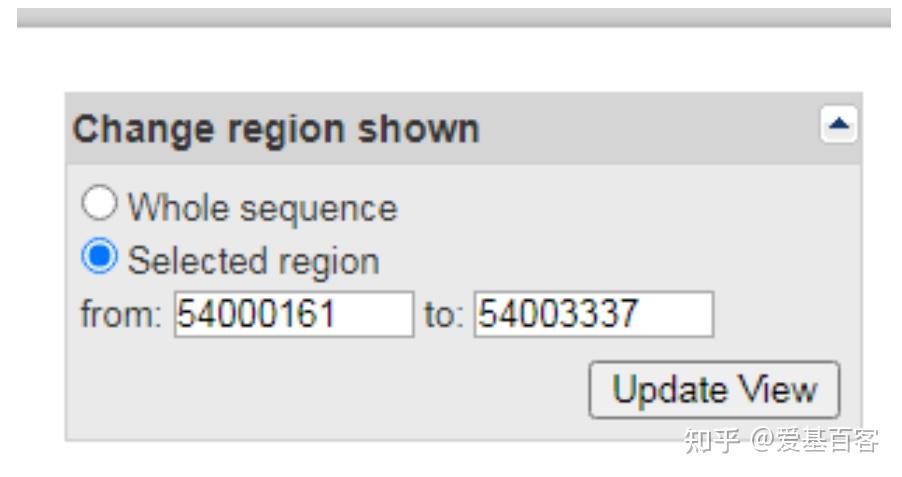
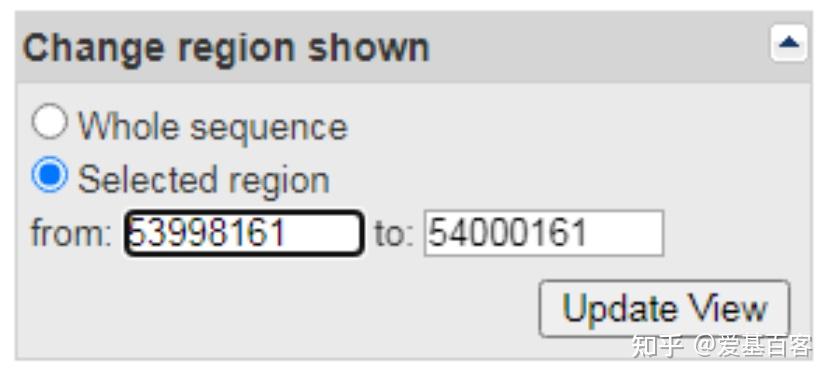
(1)点击Genomic regions, transcripts, and products一项中FASTA,查看右边Change region shown一栏。

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 






 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 发表于 2025-1-3 09:49
发表于 2025-1-3 09:49