金桔
金币
威望
贡献
回帖0
精华
在线时间 小时
|
更多内容请关注下方链接
谱度众合公众号谱度众合
前言
MaxQuant是一款免费的蛋白质组学软件包,由马克斯-普朗克生物化学研究所开发。它内置了自己的搜索引擎Andromeda,可以支持目前主流的蛋白质组学质谱仪厂商产生的原始数据格式。MaxQuant支持标记定量和非标定量,2.0版本后的MaxQuant还支持DIA的数据分析。MaxQuant具备非线性质量校正和Match Between Runs功能,可以增加蛋白鉴定数量和提高定量准确性。下面我们主要以非标定量蛋白质组学(LFQ)数据为例,向大家演示MaxQuant数据库检索的过程。
一、非标定量数据搜库过程
01 下载MaxQuant软件
进入官方网站http://www.maxquant.org 下载MaxQuant软件。软件无需安装,解压后双击MaxQuant.exe即可运行。
02 下载蛋白质序列数据库fasta文件
数据库检索(简称搜库)是将质谱采集到的原始谱图数据和蛋白质序列数据库进行比对解析的过程,因此大部分搜库软件都需要两类输入文件:蛋白质序列数据库和质谱原始数据,MaxQuant也不例外。
MaxQuant对蛋白质序列数据库fasta文件中的蛋白质序列进行模拟酶切和碎裂,将蛋白质序列信息转换为理论谱图数据。通过将理论谱图数据与实际采集到的原始谱图数据进行比对打分,实现搜库的过程。
蛋白质序列数据库fasta文件的下载方法如下:
(1)打开UniProt网站http://www.uniprot.org,以下载human蛋白质序列数据库为例,选择Taxonomy,输入Human进行搜索,点击Taxon ID 9606,进入以下页面。点击View proteomes,选择Reference proteomes。
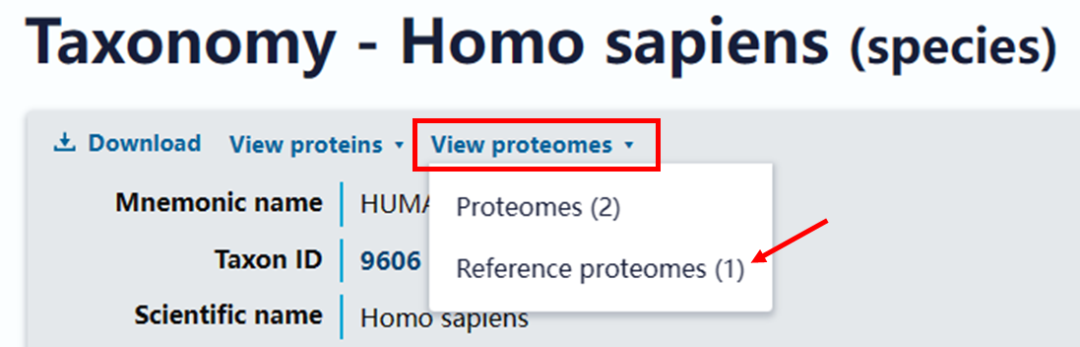
(2)在参考蛋白质组数据库页面中,点击该数据库含有的蛋白数目82685。
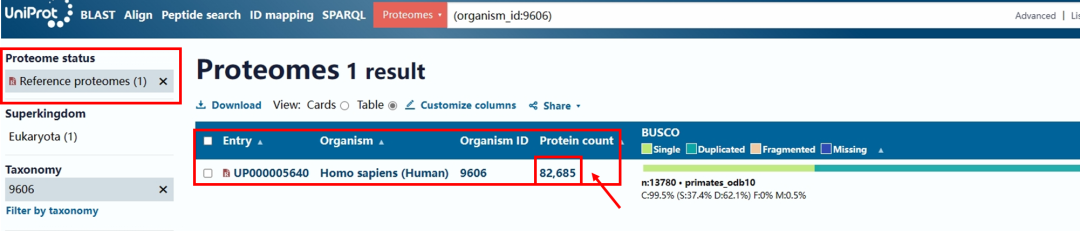
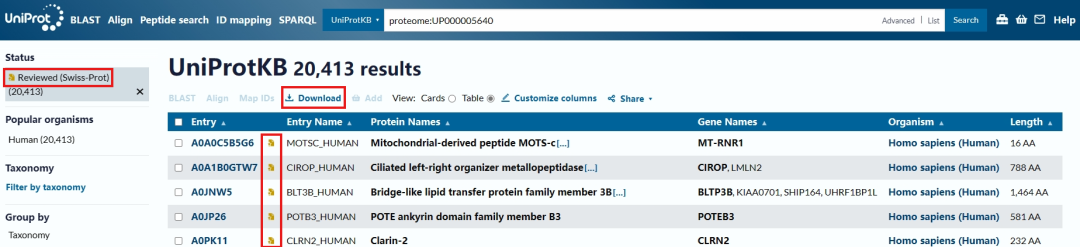
(3)跳转至新页面后,可以看到有些蛋白旁边带有黄色的图标,表示这些蛋白经过Reviewed,即人工审阅,归属于UniProt下的Swiss-Prot数据库。对于研究比较多的模式物种如人和小鼠,可选择左侧Reviewed (Swiss-Prot)的蛋白条目,点击Download,Format可选择FASTA(canonical),Compressed处选择Yes,最后点击Download下载蛋白质序列数据库fasta文件。对于其它物种,Reviewed蛋白可能偏少,一般优先选择Reference Proteome进行下载。
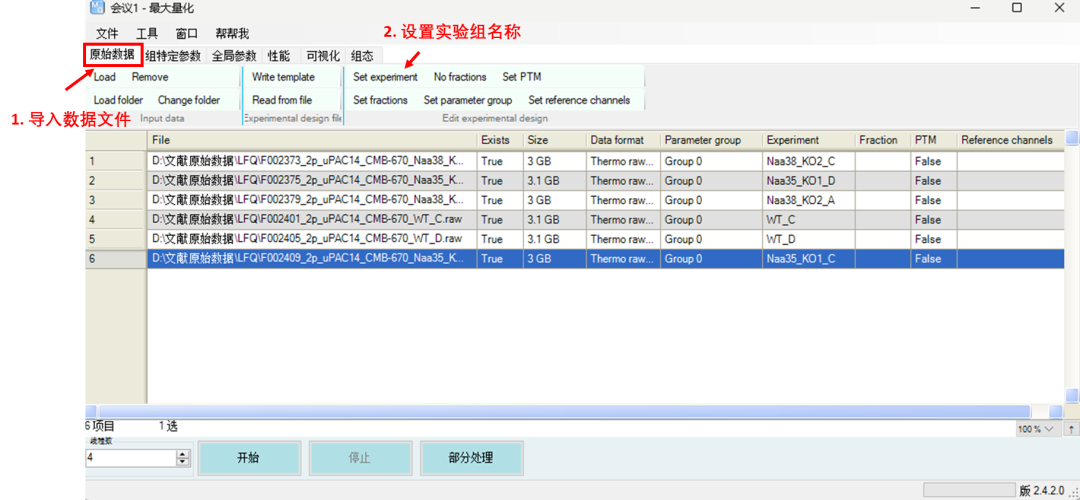
03 打开MaxQuant软件,导入质谱原始数据文件
点击Load导入数据文件,或点击Load folder导入仅包含数据文件的文件夹。目前蛋白质组学质谱仪厂商的原始数据格式有.raw (Thermo公司)、.d (Bruker公司)、.WIFF和.wiff.scan (SCIEX公司)等。可通过Set experiment设置实验组名称。这里我们用到的演示数据是从ProteomeXchange官网下载的https://proteomecentral.proteomexchange.org,蛋白质数据集为PXD034104,数据来源具体实验详见文献“N-terminal acetylation shields proteins from degradation and promotes age-dependent motility and longevity”。
04 设定组特定参数
对于常规的非标定量检测,可直接使用软件默认的参数,包括:Type——选择Standard,Modifications——选择固定修饰Carbamidomethyl (C)、可变修饰Oxidation (M)和Acetyl (Protein N-term),Digestion——选择Specific和Trypsin/P。
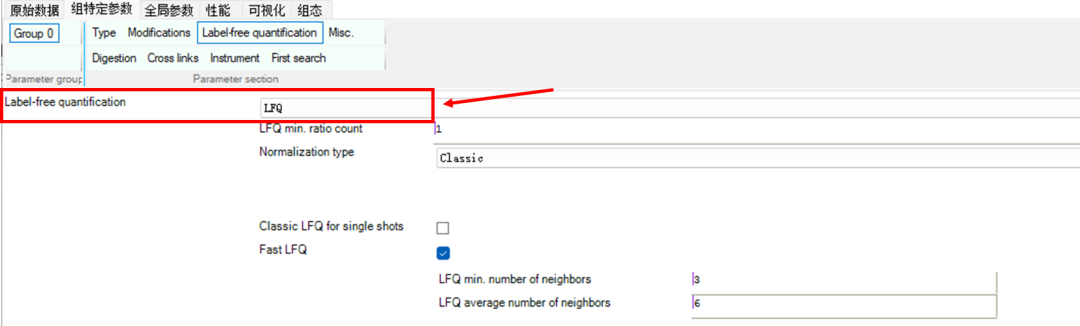
需要修改的参数:Label-freequantification——选择LFQ。
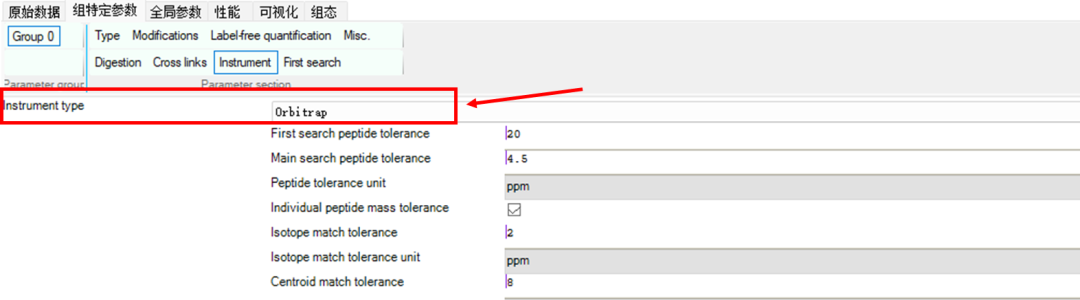
Instrument——根据实际质谱检测的仪器选择Instrument type。
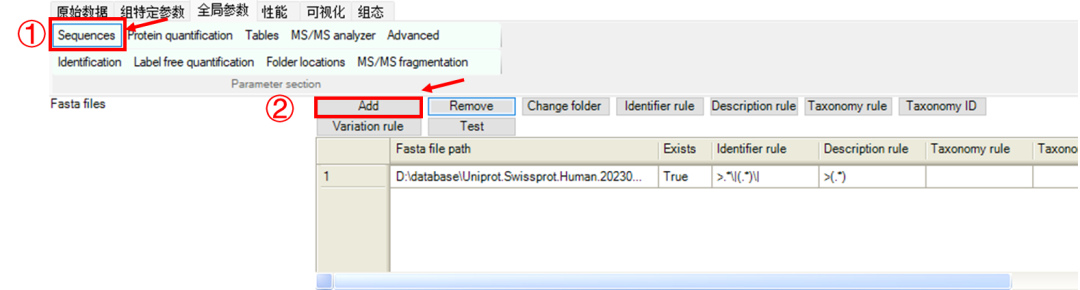
05 设定全局参数
Sequence——导入参考的Human蛋白质序列数据库fasta文件。
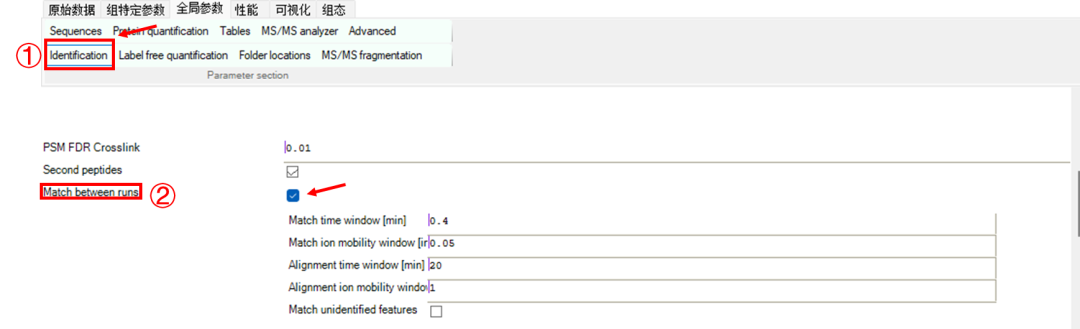
Identification——勾选Match between runs。由于缺失值的存在会削弱生物样本或实验条件之间真实定量差异的能力,Match between runs功能可以改善缺失值问题。
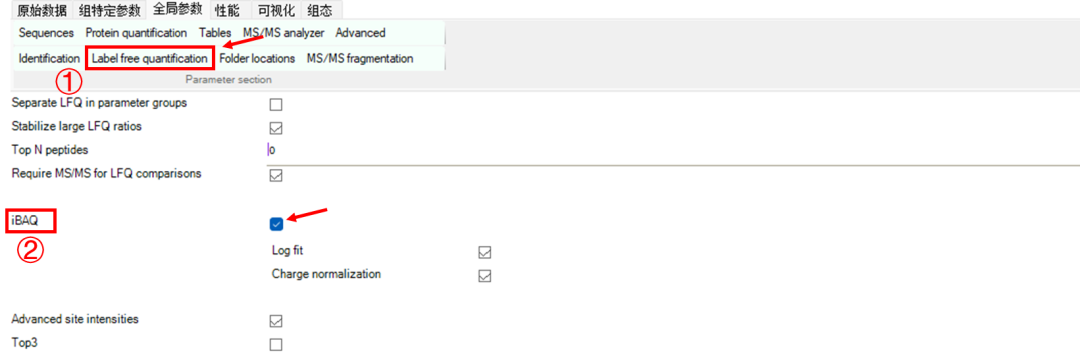
Label-free quantification——勾选iBAQ。iBAQ是基于Intensity的强度值,除以该蛋白的理论可被检测的肽段数目计算而来的定量值,主要用于不同蛋白的相互比较。
06
性能
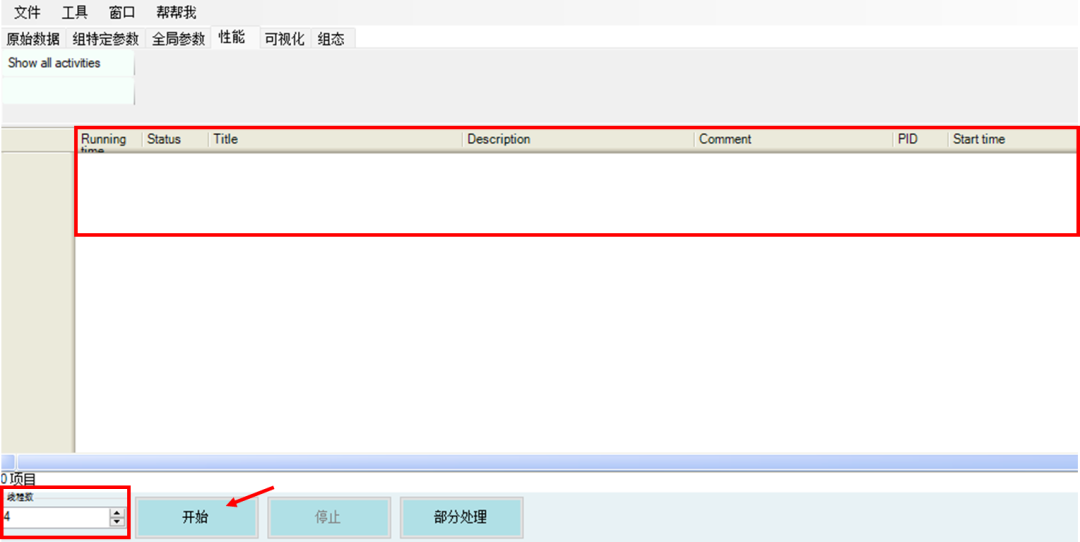
左下角线程数可根据电脑实际线程进行修改,点击开始,MaxQuant开始搜库,该页面会显示详细的搜库状态。
07 结果文件
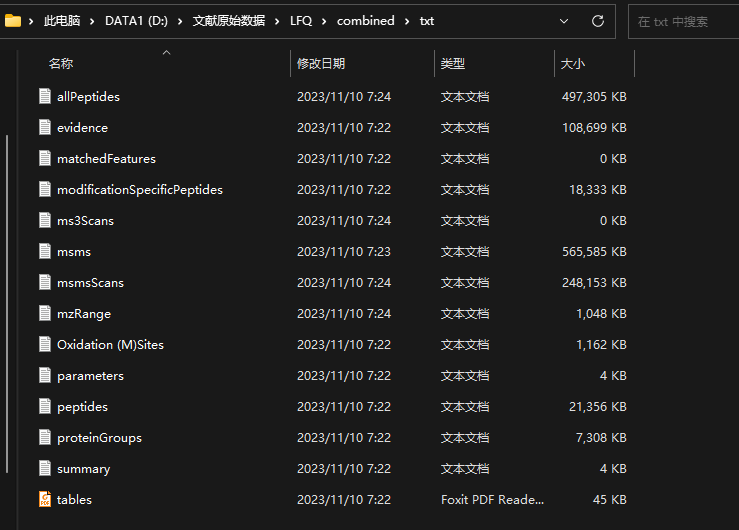
搜库完成后,主要结果在Combined文件夹下的txt子文件夹,包含下图中的文件,常用的文件有蛋白结果信息proteinGroups.txt和肽段结果信息peptides.txt。如果对于结果中的表头信息不太清楚,可查阅tables.pdf文件,其解释每个txt结果文件中各个表头的含义。
二、TMT标记定量数据搜库过程
如果是TMT标记定量的蛋白质组学数据,设置上有一些不同之处,我们这里把需要重点调整的参数拎出来,其它基本操作过程与上述步骤类似。
- 原始数据:导入质谱原始数据,如有多个组分,通过Set fractions进行设置;
- 组特定参数:此处根据标记实验和质谱采集过程选择对应参数,如Type选择Reporter MS2,Isobaric labels选择使用的标记试剂(选择后只保留使用的标记通道即可),其余参数可保持默认值;

- 全局参数:在Sequences中选择对应的fasta数据库文件,其余可保持默认值。
以上就是使用MaxQuant软件进行定量蛋白质组学数据搜库的过程啦,这一步我们只是拿到了基本的定量数据,下一篇我们继续讨论拿到MaxQuant搜库结果后该如何展开下一步的分析。小伙伴有疑问的话可以在文章下留言哦!
参考资料
1.Tyanova, S., Temu, T. & Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nature Protocols 11, 2301–2319 (2016).
2.Sinitcyn, P., et al., MaxDIA enables library-based and library-free data-independent acquisition proteomics. Nature Biotechnology 39, 1563-1573 (2021).
3.UniProt数据库网站http://www.uniprot.org
4.Varland, Sylvia et al. N-terminal acetylation shields proteins from degradation and promotes age-dependent motility and longevity. Nature communications 14(1), 6774 (2023).
 |
|

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 






 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 发表于 2024-9-14 14:08
发表于 2024-9-14 14:08





