登陆有奖并可浏览互动!
您需要 登录 才可以下载或查看,没有账号?立即注册


×
急性感染后,CD8+ T细胞增殖并分化为细胞毒性T淋巴细胞(ctl)以消除感染的靶细胞。ctl的分化与转录、表观遗传和代谢重组相平行,以支持其克隆扩增和效应功能,如细胞因子分泌和细胞毒性。在病原体被清除和炎症消退后,大多数效应T细胞死亡,只有少数淋巴细胞作为长期记忆T细胞保留下来。
然而,当抗原不能被消除时,例如在持续性感染和癌症中,持续的抗原受体刺激会驱动另一种分化轨迹,即T细胞耗竭。耗竭的T (Tex)细胞在表型上表现为PD-1、Tim-3和Lag-3等共抑制受体的上调,在功能上表现为细胞因子分泌和细胞溶解活性的下降。尽管在终末分化过程中,特克斯细胞会失去增殖能力并发生凋亡,但它们会被一个独特的多能细胞群体稳定地补充,这些细胞被称为耗尽的T (Tpex)细胞的前体细胞(或祖细胞)。Tpex细胞可以通过表达记忆相关分子,如转录因子TCF-1和细胞表面分子Ly108和CD62L来识别,但没有终端分化标记,如Tim-3和CD160。Tpex细胞的“干样”自我更新能力及其在慢性感染小鼠的过继转移后优越的再生潜力激发了越来越多的科学兴趣,以确定控制从多能Tpex向功能失调的Tex细胞转变的分子机制。这个问题也与临床相关,因为Tpex细胞是治疗性检查点阻断后抗肿瘤免疫反应的主要介质。虽然PD-1在Tpex和终末耗竭的T细胞上都有表达,但只有Tpex细胞对抗PD-1治疗有有效的反应,表现为增殖爆发和分化为细胞毒性T细胞,从而在接受检查点免疫治疗的患者中重新激活抗肿瘤免疫反应。然而,维持Tpex细胞干性的分子机制和指示其功能衰竭的信号仍不完全清楚,值得进一步研究以改善癌症免疫治疗的临床结果。
研究概要
T细胞衰竭是癌症和持续性感染的标志,其特征是抑制性受体上调、细胞因子分泌减少和细胞溶解活性受损。最终耗竭的T细胞由前体细胞群(Tpex)稳定地补充,但是控制Tpex维持的代谢原理和控制其耗竭的调节回路仍然不完全清楚。通过结合基因缺陷小鼠、单细胞转录组学和代谢组学分析,我们发现线粒体功能不足是启动T细胞功能衰竭的细胞内在触发因素。在分子水平上,我们发现线粒体功能障碍引起氧化还原应激,从而抑制缺氧诱导因子1α (HIF-1α)的蛋白酶体降解,并促进Tpex细胞的转录和代谢重编程,使其成为终衰竭的T细胞。我们的发现也具有临床意义,因为嵌合抗原受体(CAR) T细胞的代谢工程是一种很有前途的策略,可以增强肿瘤免疫治疗中Tpex细胞的干性和功能。
结果
(1)终端T细胞耗竭以代谢重编程为特征
为了分析枯竭T细胞沿发育轨迹的转录代谢程序,我们对感染淋巴细胞性脉络丛脑膜炎病毒(LCMVCL13)克隆13株后从慢性感染野生型(WT)小鼠中分离的CD44+ PD-1hi CD8+ T细胞进行了单细胞(sc) RNA测序(补充图1a)。为了避免由于活跃细胞分裂造成的偏差,我们专注于非增殖T细胞,并使用均匀流形近似和投影(UMAP)可视化的无监督聚类识别出六个主要的耗尽T细胞群体(图1a和补充图1b)。Tpex细胞的特征是活化(Cd44, Cd69, Icos),耗竭(Pdcd1, Tox)和记忆相关分子(Sell, Ccr7, Tcf7, Slamf6, Id3, Cxcr5)的独特组合(图1b-d和补充图1b, c)。扩散伪时间分析预测了源于“茎样”CD62L+ Tpex1细胞的两条分化轨迹(图1b)。其中一条轨迹合并为“类似ctl”的耗尽T细胞群,其特征是表达Cx3cr1、Klrg1和S100a4(补充图1c)26。第二个谱系指向最终耗尽的T细胞(图1b)。事实上,Tex1和Tex2亚群显示出终端衰竭标记的渐进式上调,如Havcr2(编码Tim-3)、Cxcr6、Id2、Lag3、Gzma、Cd244a (2B4)和Tnfrsf9 (4-1BB)(图1c、d和补充图1c)。这两个谱系的分叉发生在小而不同的Ttransit集群中,该集群的标志是tpx相关基因的下调,包括Sell (CD62L)、Ccr7、Tcf7、Il7r、Id3和Slamf6 (Ly108),以及末端耗尽标记物的中间表达(图1d)。为了进一步探索干细胞样CD62L+ Tpex细胞向ctl样和终末耗散T细胞分化过程中的代谢转录程序,我们计算了糖酵解、线粒体呼吸、脂质代谢和戊糖磷酸途径等代谢途径的基因集富集分数(Supplementary Table S1)。这些分析显示,在Tpex向终末分化T细胞过渡时,线粒体(基因集ID M9577)和呼吸链基因(M19046)表达急剧下降(图1e),表明线粒体(dys-)功能与T细胞衰竭之间存在关系。与我们的scRNA测序数据并行,我们分析了来自慢性感染小鼠的ID3+ Tpex和Tim-3+ Tex细胞的公开大量RNA测序数据4(补充图1d, e)。基因集富集分析(GSEA)和基因网络聚类揭示了线粒体代谢和翻译途径与Tpex细胞的显著相关性,而Tex细胞的转录组主要与信号转导相关。细胞周期和DNA修复基因表达特征。GSEA利用基因本体(GO)途径,如线粒体基因(M9577)、线粒体翻译(M27446)以及通过化学渗透偶联(M1025)进行呼吸电子传递和ATP合成,进一步支持了Tpex细胞比Tex细胞更依赖线粒体呼吸的观点。
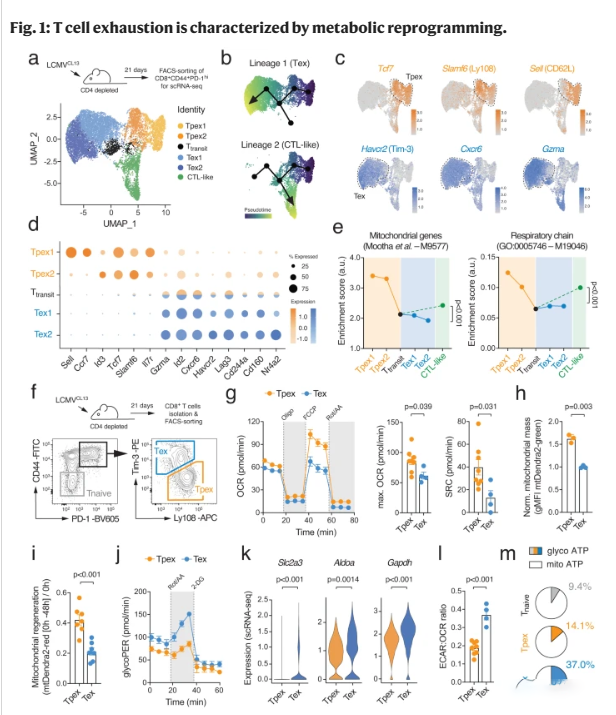
为了直接测量耗尽T细胞的线粒体呼吸,我们用LCMVCL13感染WT小鼠,并通过FACS分选分离Ly108+ Tpex和Tim-3+ Tex细胞(图1f)。与转录组学数据一致,细胞外通量分析显示,与Tex细胞相比,Tpex的基础和最大耗氧率(OCR)更高(图1g),表明Tpex细胞更依赖于OXPHOS。以最大OCR和基础OCR之差计算的备用呼吸能力(SRC)表明,Tpex细胞具有更大的代谢储备来利用线粒体呼吸,而Tex细胞几乎以最大容量操作OXPHOS(图1g)。SRC的差异可以通过线粒体含量的改变和/或线粒体再生能力的差异来解释。为了测试这些可能性,我们使用mitto -Dendra2小鼠27,它们表达线粒体定位版本的Dendra2蛋白。利用与线粒体质量/体积相关的mito-Dendra2的绿色荧光27,我们可以很容易地检测到Tpex细胞的线粒体含量明显高于Tex细胞。为了描述耗尽T细胞的线粒体更新和再生能力,我们用细胞微量紫(CTV)标记慢性感染mito-Dendra2小鼠的T细胞,并在405 nm激光照射下不可逆地将线粒体荧光发射光谱从绿色荧光切换为红色荧光.我们在体外追踪了未分裂的Tpex和Tex细胞中非红色线粒体的出现,作为线粒体再生的一种措施。Mitotracker和TMRE探针证实,与Tpex细胞相比,Tex细胞的线粒体含量和膜电位更低。此外,Tex细胞显示电子传递链(ETC)复合物和线粒体生物发生因子,如PGC-1α和TFAM 的表达减少。
与其线粒体功能受损相比,与Tpex细胞相比,Tex细胞表现出糖酵解质子外排率(glycoPER)升高(图1j),这表明Tex细胞通过有氧糖酵解升高来补偿线粒体功能不足。Tex细胞细胞外酸化率(ECAR)的增加与营养转运蛋白和糖酵解酶如Slc2a3(编码GLUT328)、Aldoa和Gapdh的表达增加相关(图1k)。与前体细胞相比,特克斯细胞的ECAR:OCR比值更高,这进一步支持了T细胞衰竭是由线粒体呼吸到有氧糖酵解的代谢转换介导的这一观点(图11)。当我们计算糖酵解和线粒体代谢对ATP产生的贡献时,我们发现Tpex细胞主要依赖于OXPHOS,而Tex细胞对有氧糖酵解的依赖性要高2.5倍(图1m)。总的来说,这些数据表明线粒体呼吸减少和类似Warburg效应的糖酵解重编程是晚期T细胞衰竭的代谢标志。
(2)线粒体呼吸控制T细胞的功能性衰竭
已经确定线粒体ATP生成受损与Tpex向Tex细胞的转变是一致的,我们开始描述这些代谢变化是T细胞衰竭的结果还是原因。
(3)氧化应激稳定Tpex细胞中的HIF-1α蛋白水平
为了了解mpc缺陷T细胞的代谢变化如何控制T细胞衰竭过程中的复杂转录过程,我们使用液相色谱和质谱(LC/MS)分析了它们的代谢组(图4a)。mpc缺陷T细胞中下调最多的代谢物是几种糖酵解中间体,如果糖-6-磷酸、果糖-1,6-二磷酸、二羟丙酮磷酸(DHAP)、甘油醛-3-磷酸(G3P)和磷酸烯醇丙酮酸(PEP),这可能是由于糖酵解通量加速所致。此外,在mpc缺失的T细胞中,烟酰胺腺嘌呤二核苷酸磷酸(NADPH)的还原形式下调(图4a),导至NADPH/NADP+平衡移位(图4b)。
总的来说,这些数据表明氧化应激和线粒体功能不足引起的代谢重编程支持HIF-1α蛋白稳定和T细胞衰竭。
(4)hif -1α介导的糖酵解重编程支持终端T细胞分化
接下来,我们研究了HIF-1α消融如何影响慢性病毒感染期间的终端t细胞衰竭。总之,这些发现表明,线粒体功能不全和hif -1α介导的糖酵解重编程都会导至T细胞衰竭,而糖酵解重编程的药理抑制是一种可行的代谢干预策略,可以在慢性病毒感染和癌症免疫治疗期间维持(CAR) T细胞的干性、寿命和功能。
| 
 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 





 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 雷达卡
雷达卡 发表于 2023-12-21 17:07
发表于 2023-12-21 17:07
 提升卡
提升卡


