只需一步,快速开始
微信扫一扫,快速登录
您需要 登录 才可以下载或查看,没有账号?立即注册
使用道具 举报
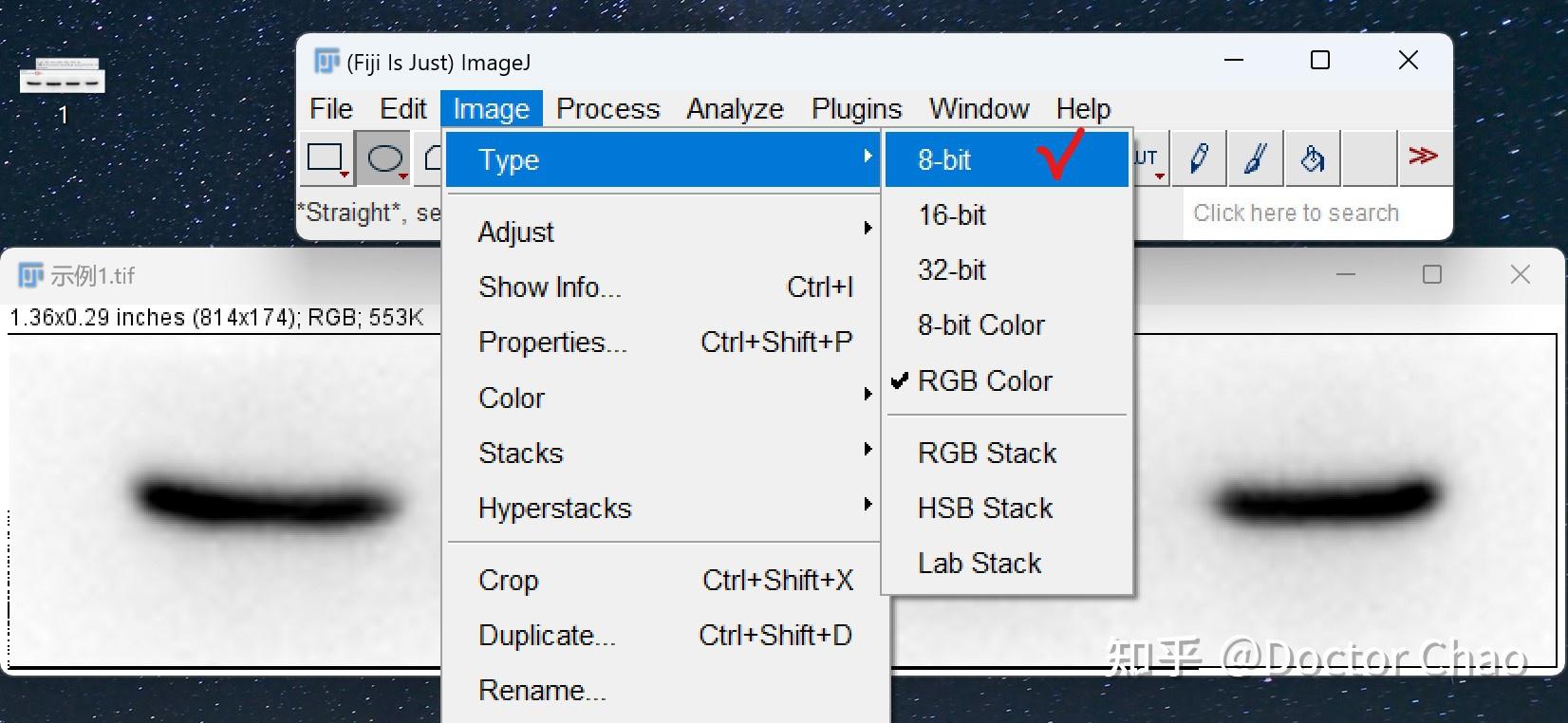
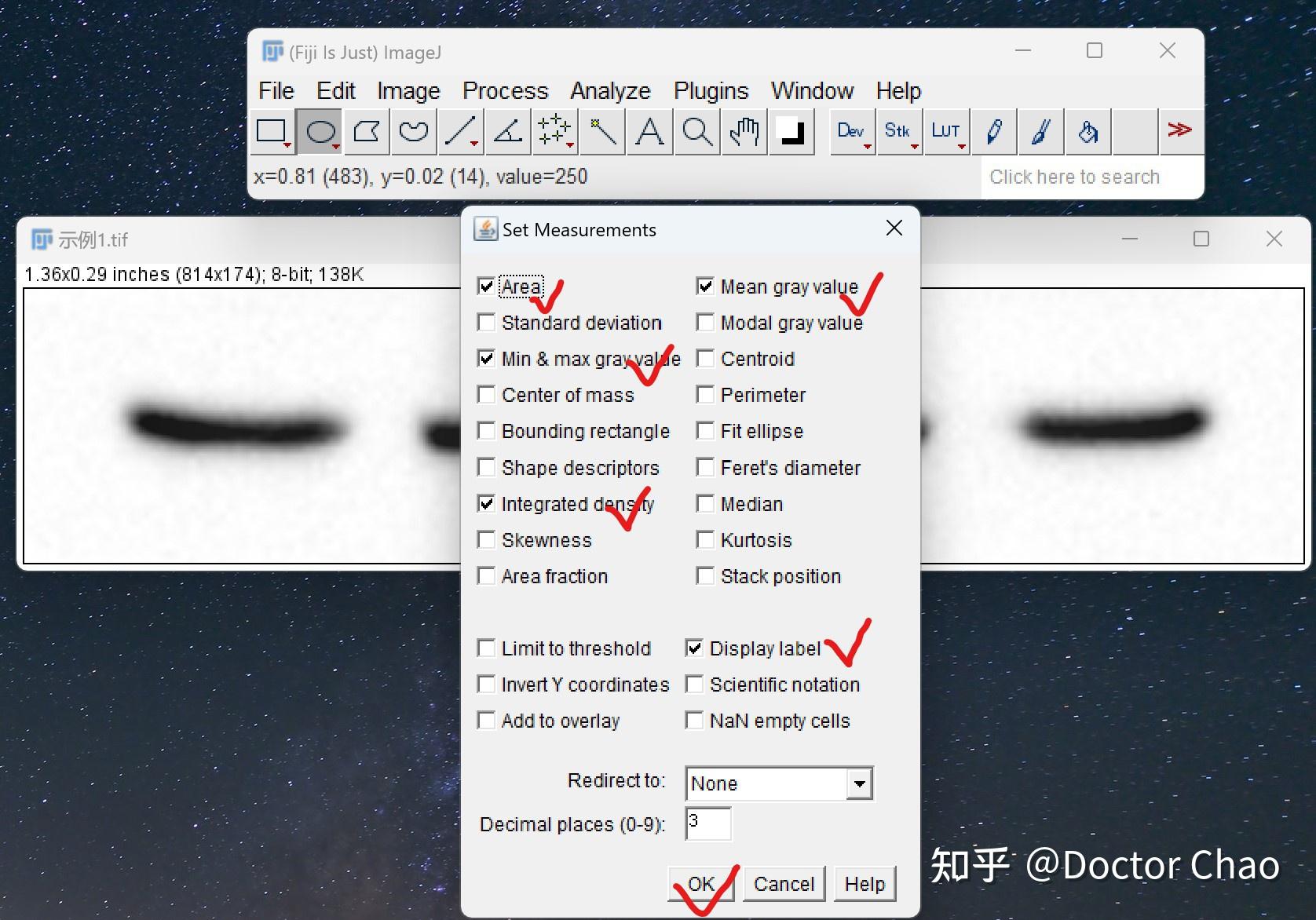
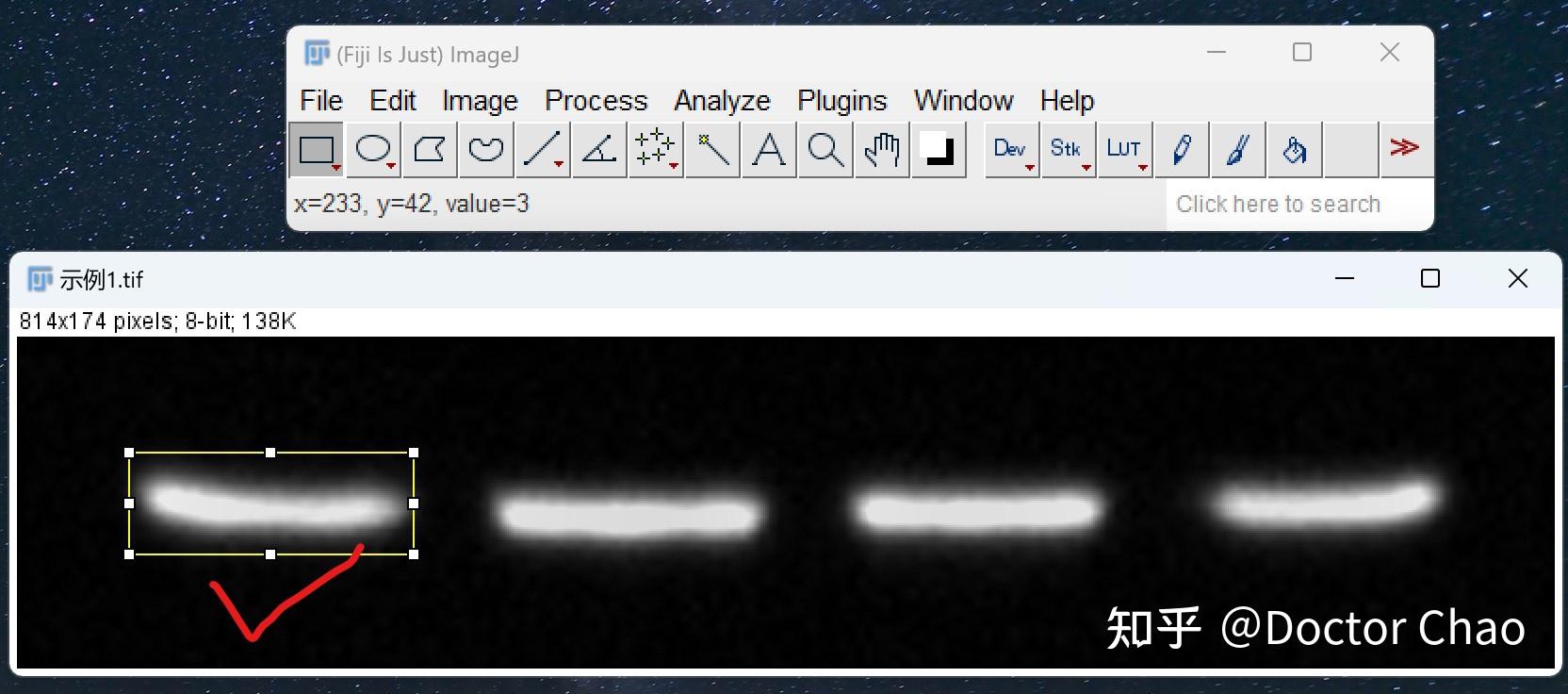
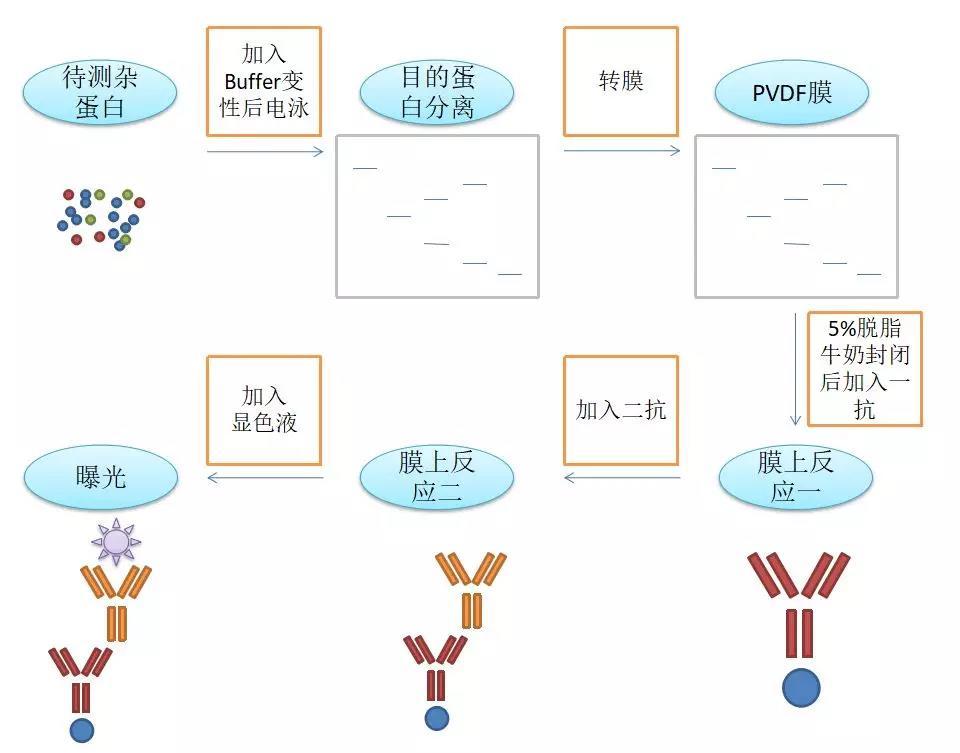
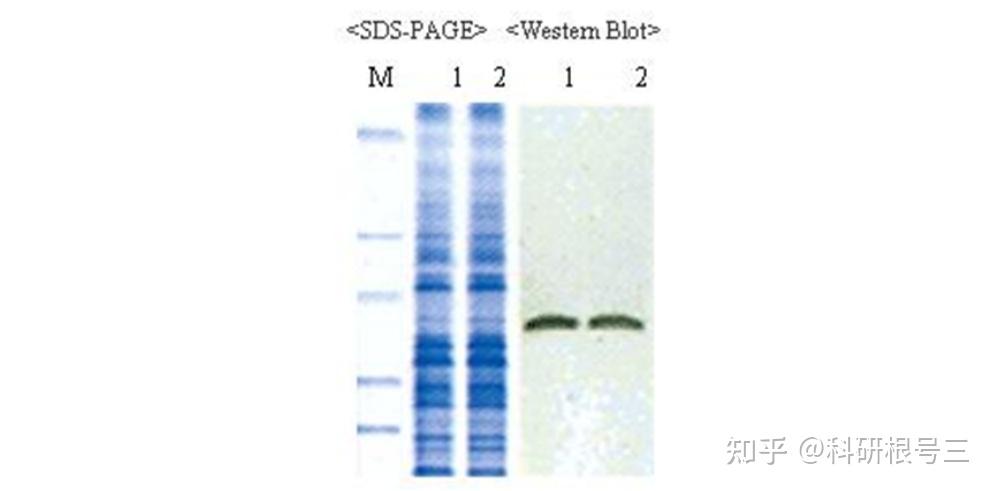
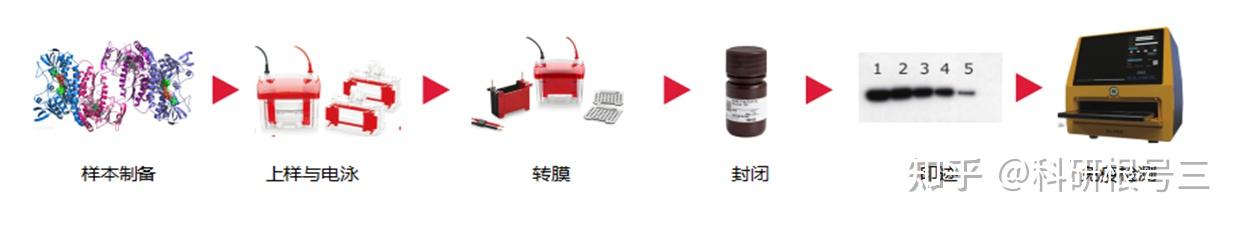
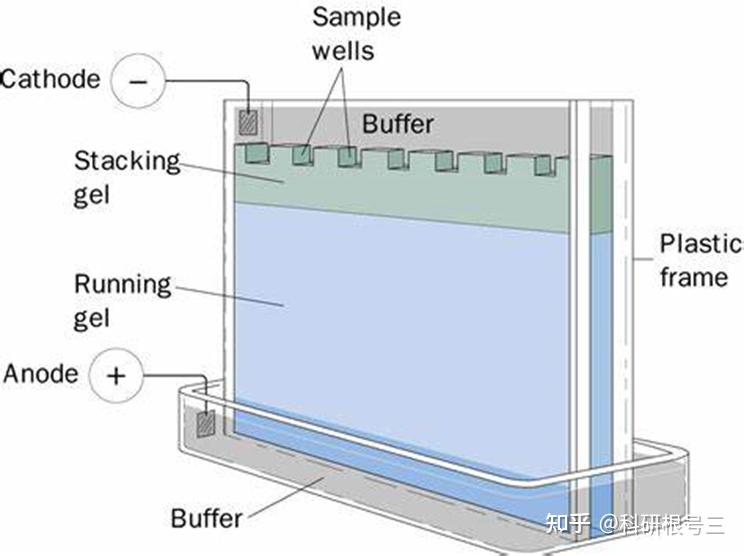
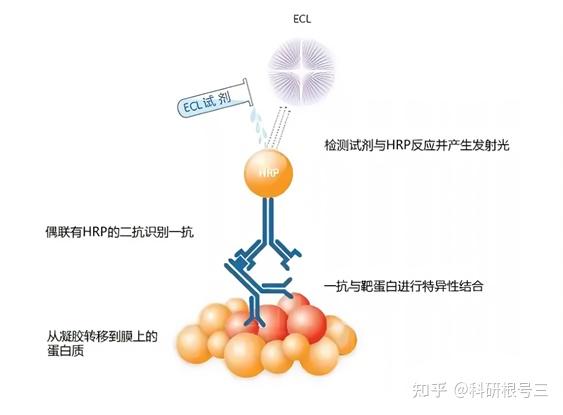
原理:通过电泳区分不同的蛋白组分,并转移至固相支持物,通过特异性抗体作为探针,对靶蛋白进行检测。
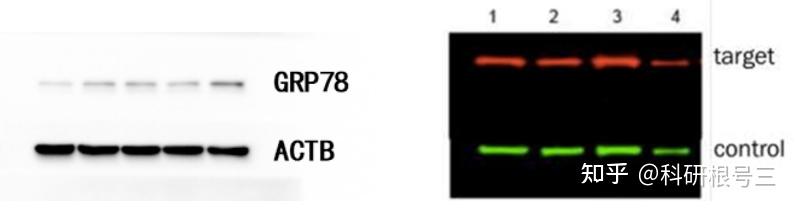
用途:定性检测,判断目标蛋白有无表达,目标蛋白分子量大小。常配合ELISA、FACS、IHC、IF等手段使用。
最全的生物实验protocol与生物实验操作视频学习资料 112个科研软件免费版(数据处理+绘图+翻译+生物实验相关等) 下载链接:https://pan.quark.cn/s/34a75dd2241d
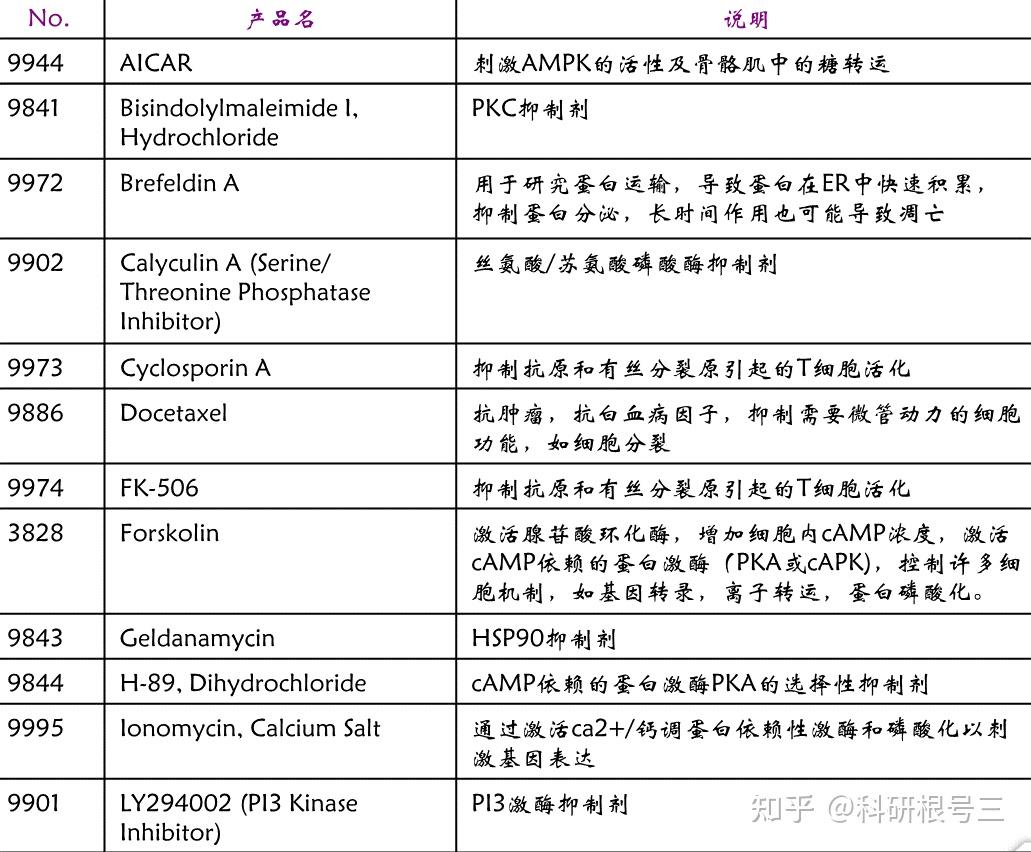
Uniprot:http://biogps.org/#goto=welcome 解靶标蛋白基因信息、别称、功能、可变剪切、定位及可修饰类型等 Proteinatlas:https://www.proteinatlas.org/ 通过IHC提供基因在正常组织和肿瘤组织中的表达变化和定位 BioGPS:http://biogps.org/#goto=welcome 不同样本中基因的mRNA表达水平和靶标蛋白的表达水平 PAXdb:http://pax-db.org/ 不同物种组织和细胞中蛋白丰度 DepMap:https://depmap.org/portal/ 肿瘤细胞mRNA表达量
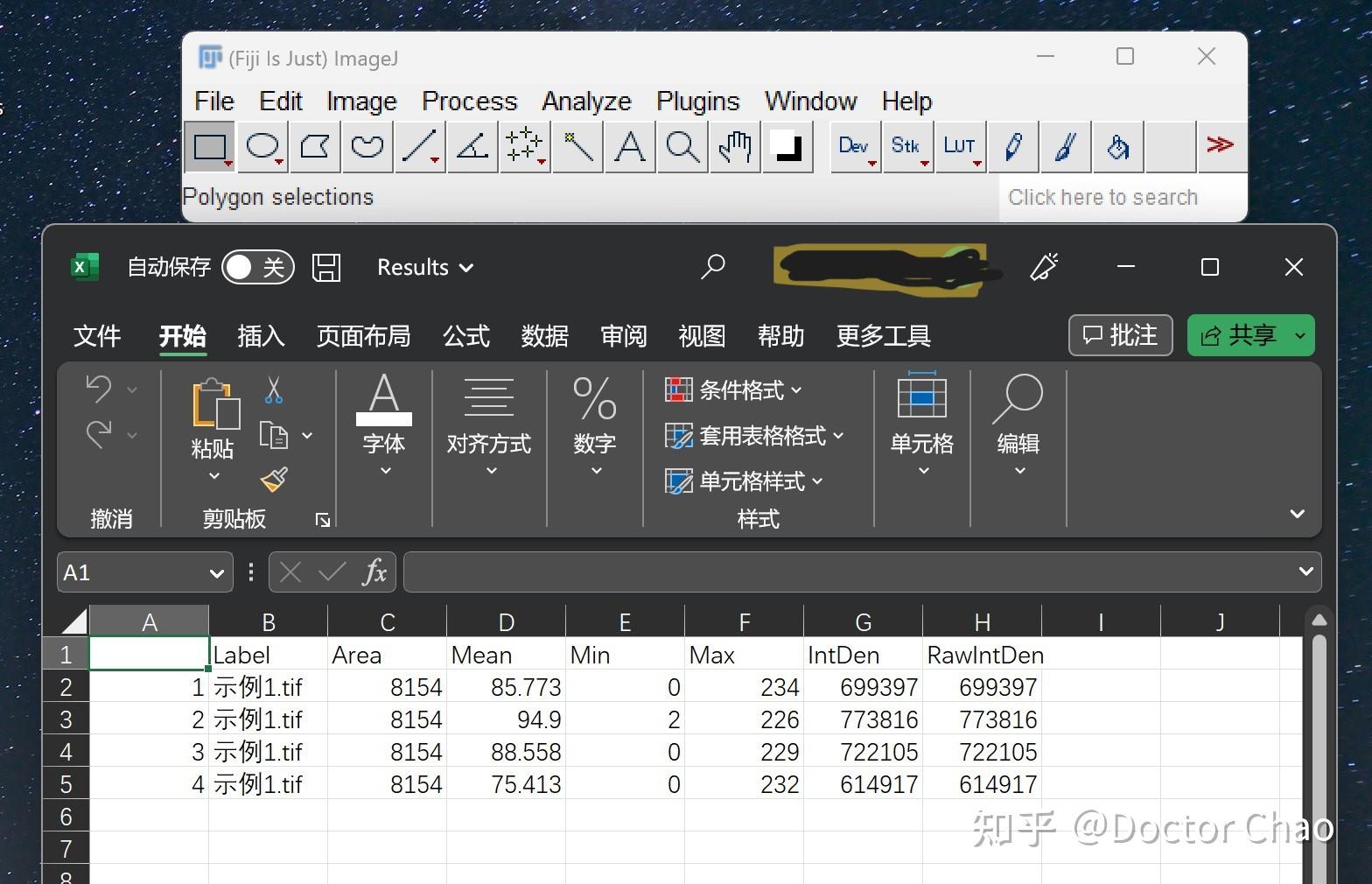
参考文献 产品说明书 预实验
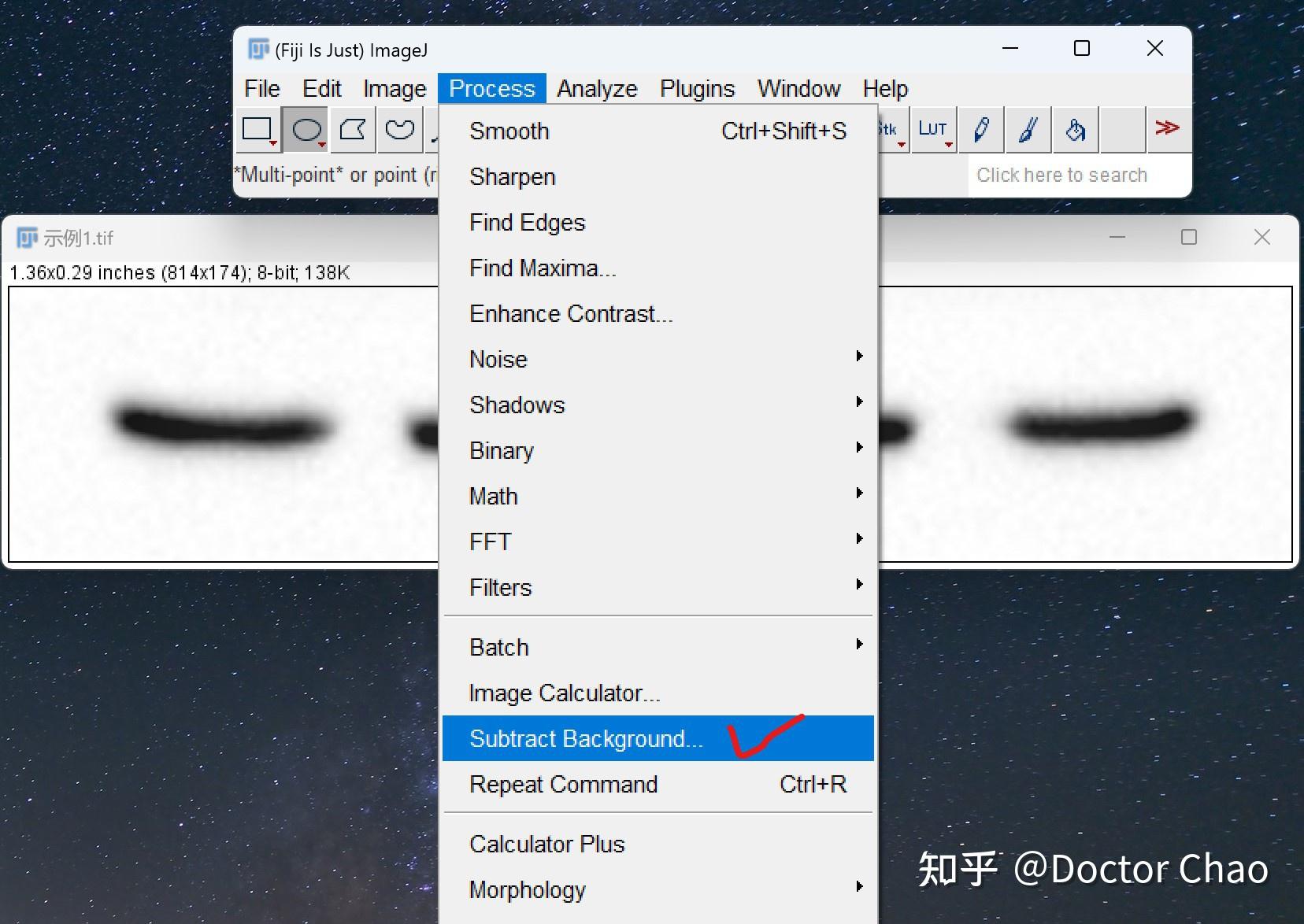
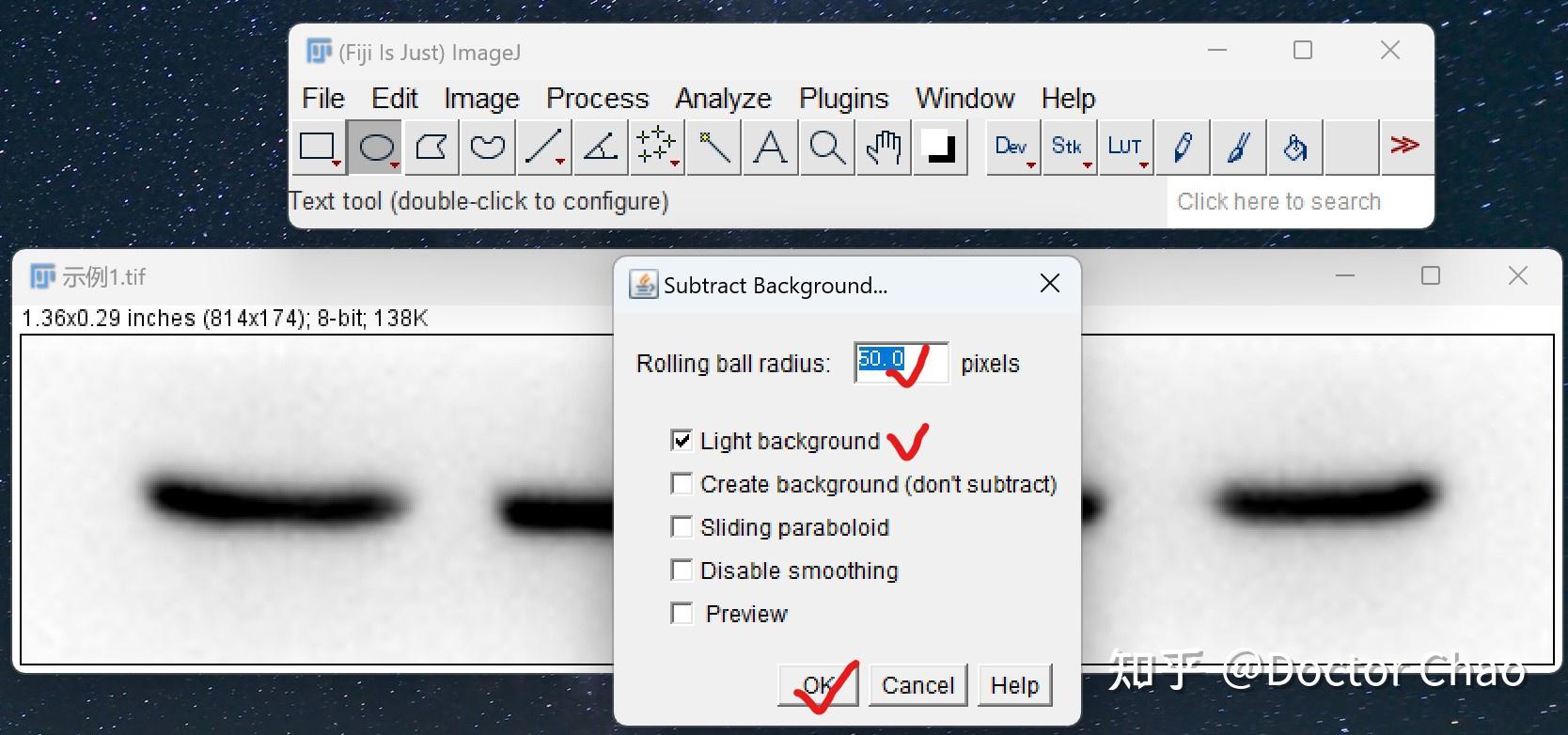
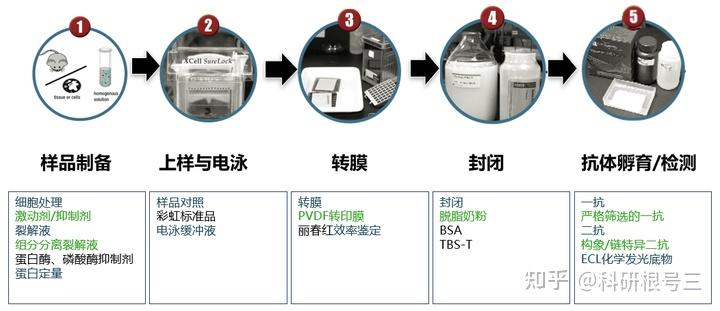
内源性和外源性的蛋白酶会在细胞或组织裂解后迅速降解蛋白。 在细胞裂解的步骤加入蛋白酶抑制剂混合物可确保蛋白免于降解,磷酸酶抑制剂可以保持蛋白的磷酸化状态。
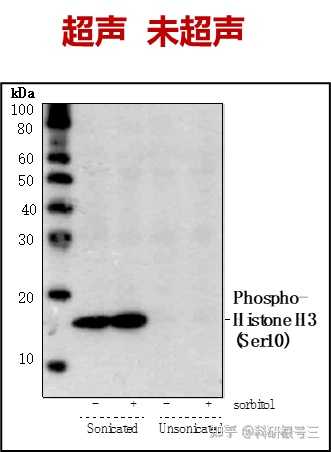
超声去除裂解样品中DNA干扰,经过超声处理过的样本得到的结果更清晰。
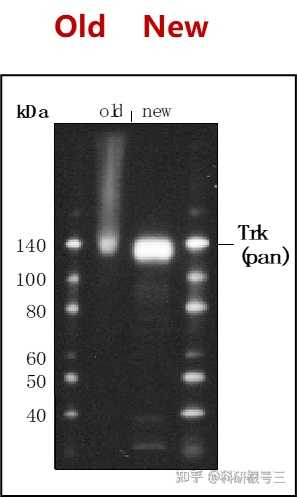
尽量使用新鲜的样品,新鲜的样品制备提取物背景较低。
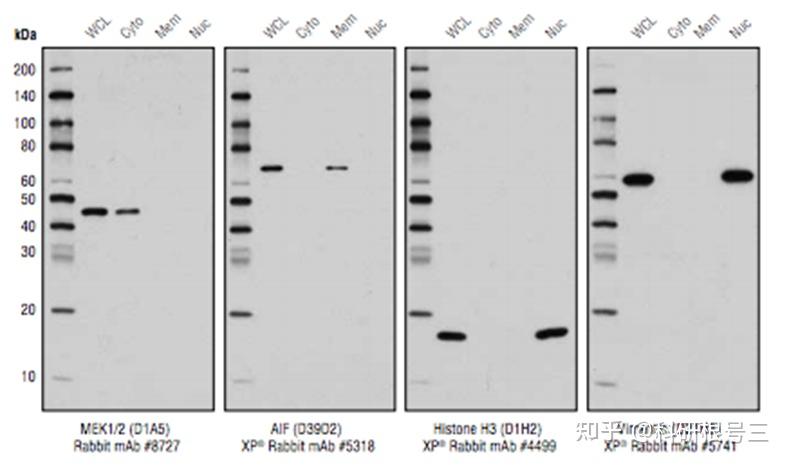
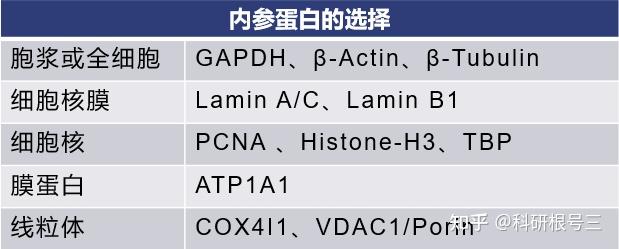
产品推荐:Cell Fractionation Kit #9038
产品推荐:Cell Fractionation Antibody Sampler Kit #11843
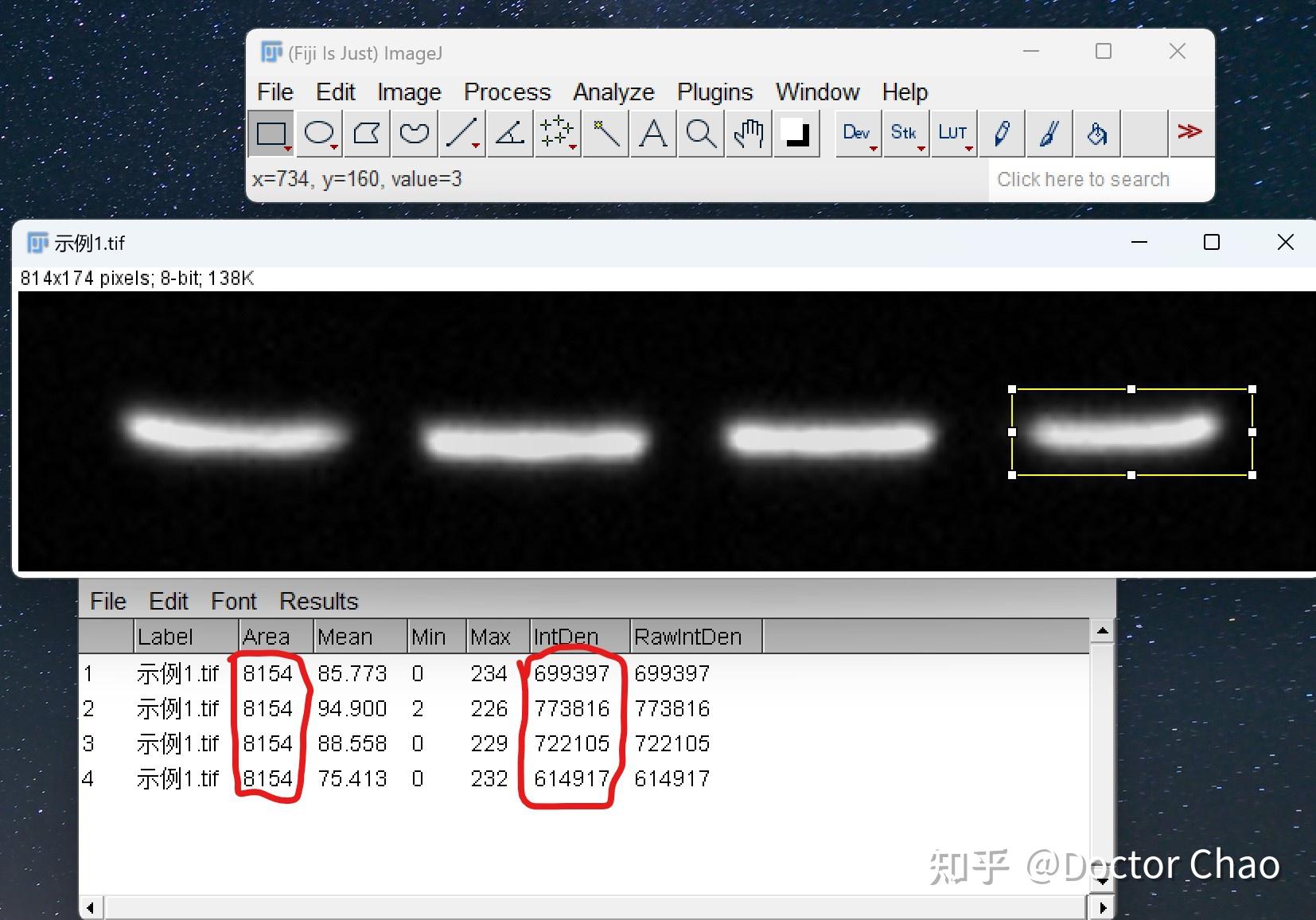
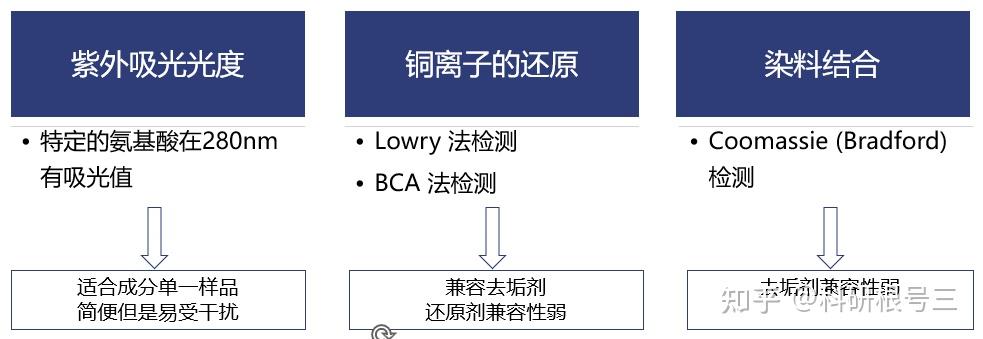
产品推荐:BCA Protein Assay Kit #FMS-W-002 产品推荐:考马斯亮蓝(Bradford)法蛋白定量试剂盒 #FMS-W-001
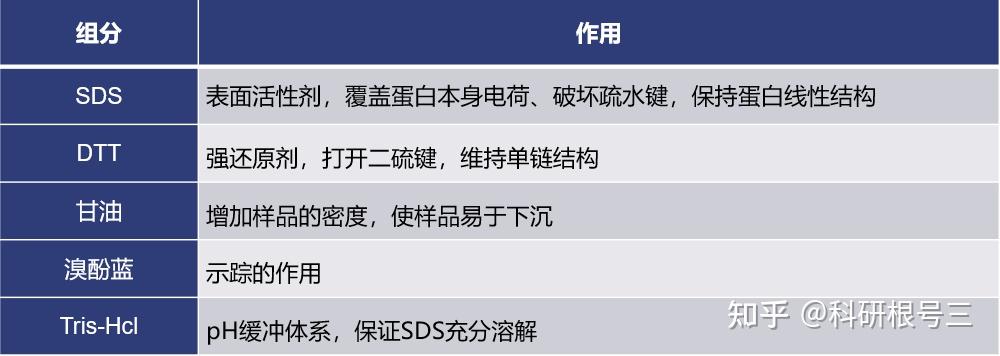
在loading buffer 中加入还原剂,如beta 巯基乙醇,不要煮沸,使用低于70°C的温度,孵育30-60 min。
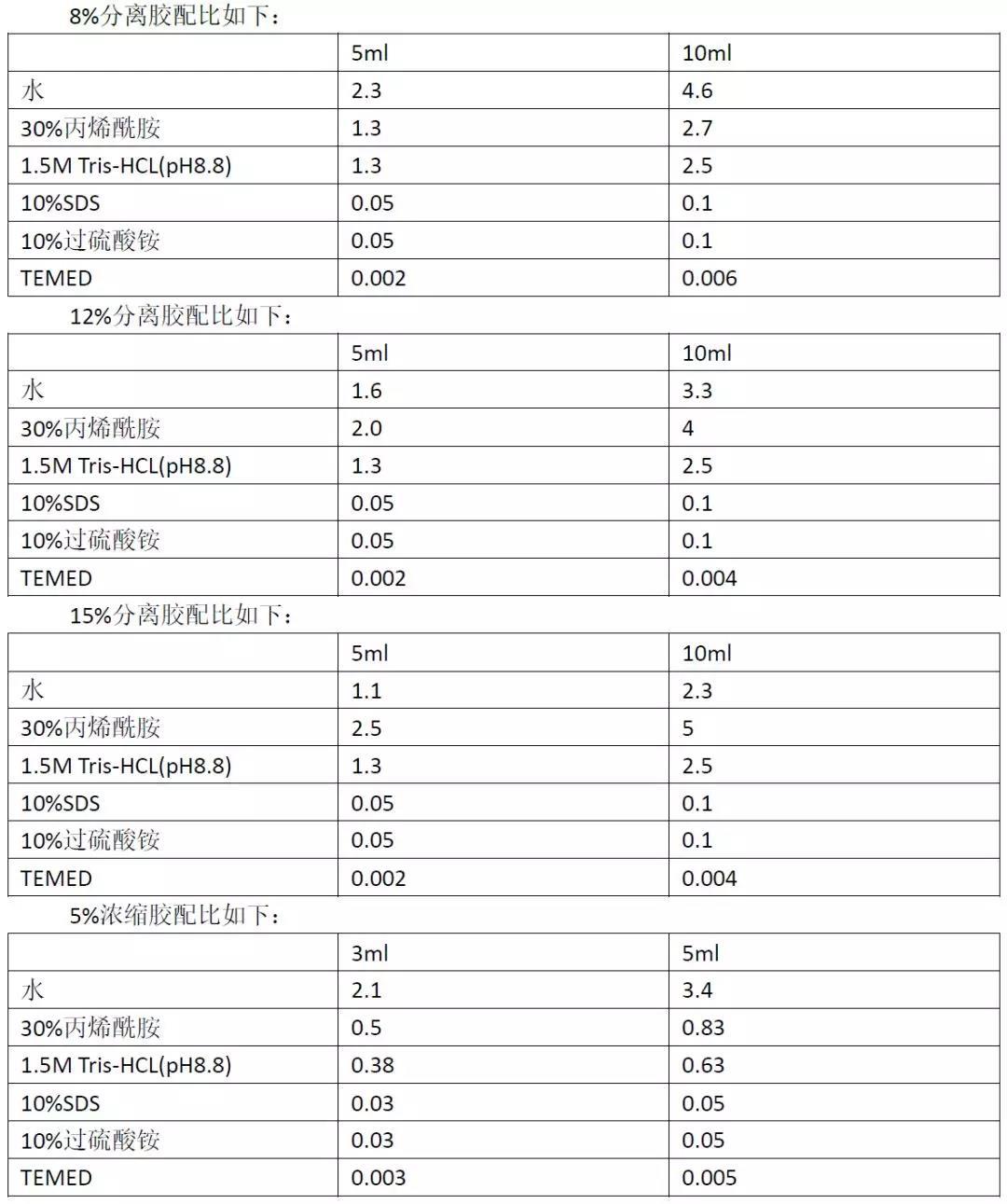
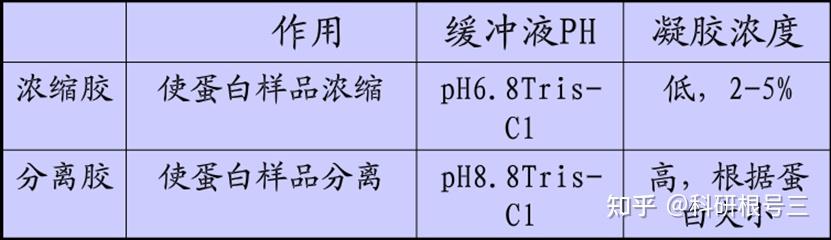
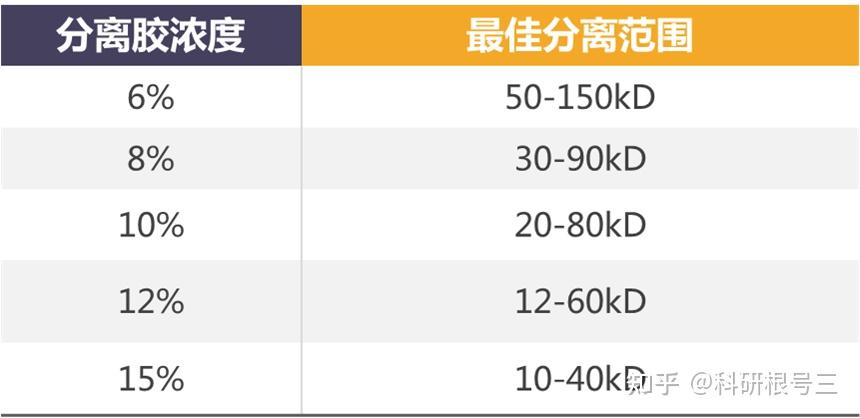
高比例的凝胶具有较小的孔径,用于分离分子量较低的蛋白。 低比例的凝胶具有较大的孔径,用于分离分子量较大的蛋白。
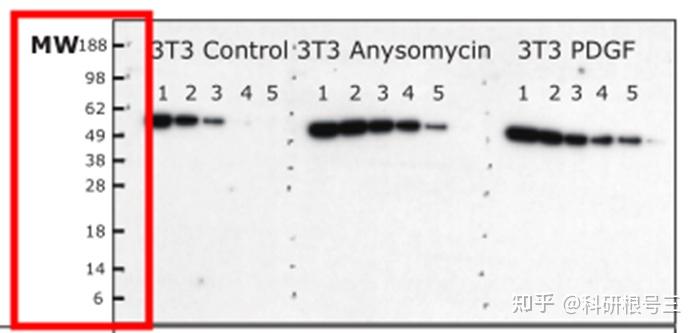
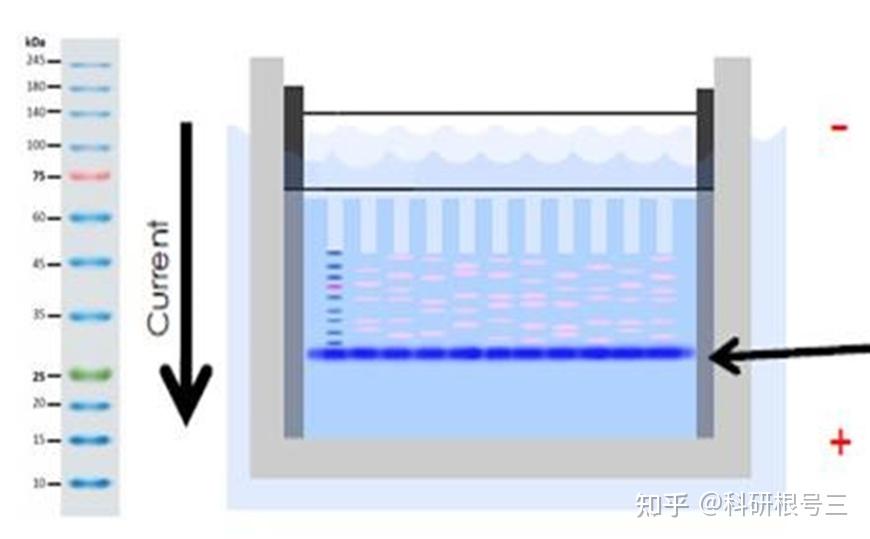
产品推荐:彩虹预染marker (5-245 kDa) #FMS-WB003
产品推荐:Tris-Glycine SDS Running Buffer (10X) #FMS-WB032
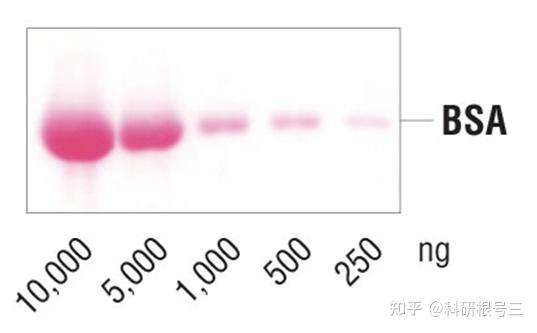
CST相关产品推荐:Ponceau S Staining Solution #59803
化学发光法:含5% 脱脂奶粉的TBST 荧光法:含5% 脱脂奶粉的TBS
脱脂奶粉 #FMS-WB020; BSA #FMS-WB021; Animal-Free Blocking Solution (5X) #15019
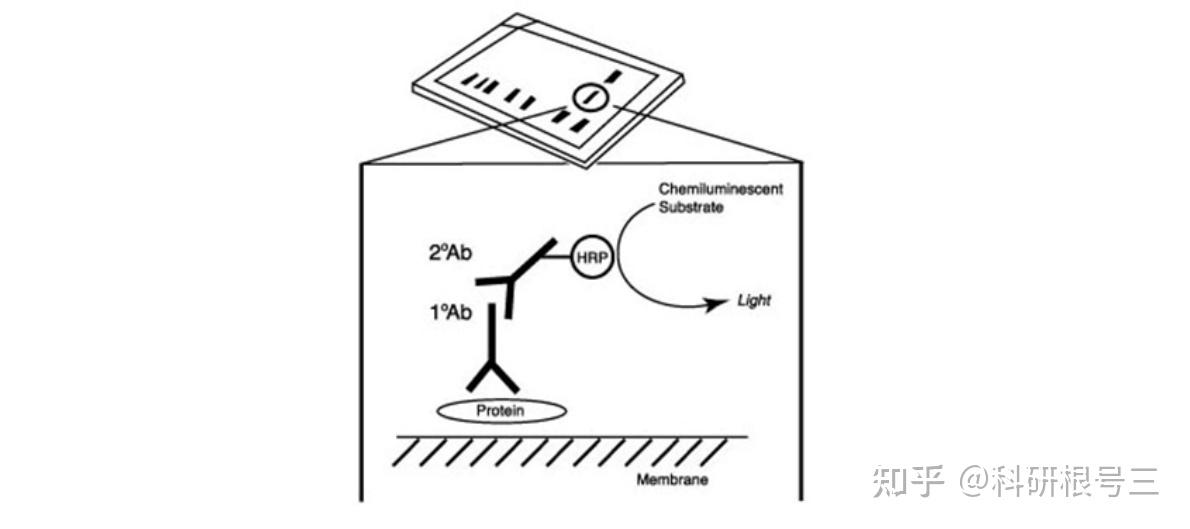
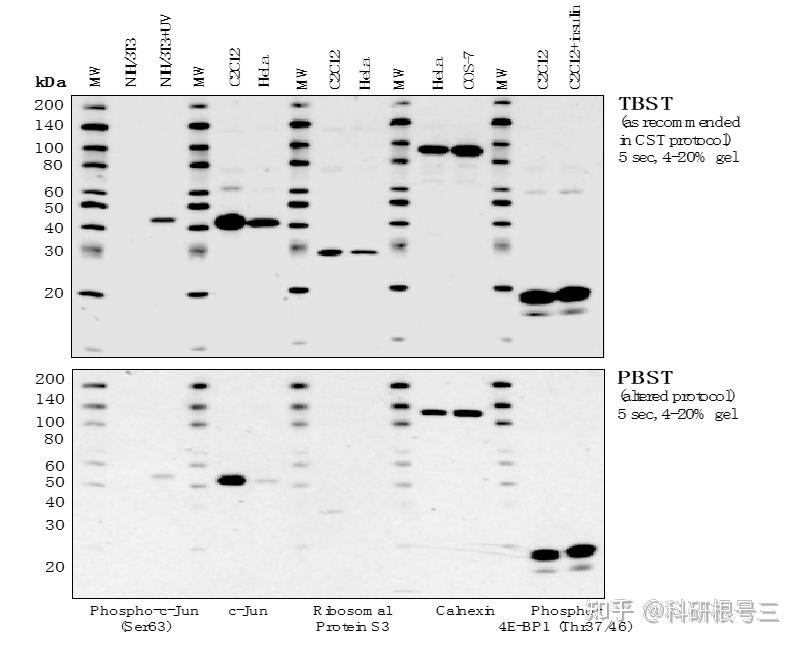
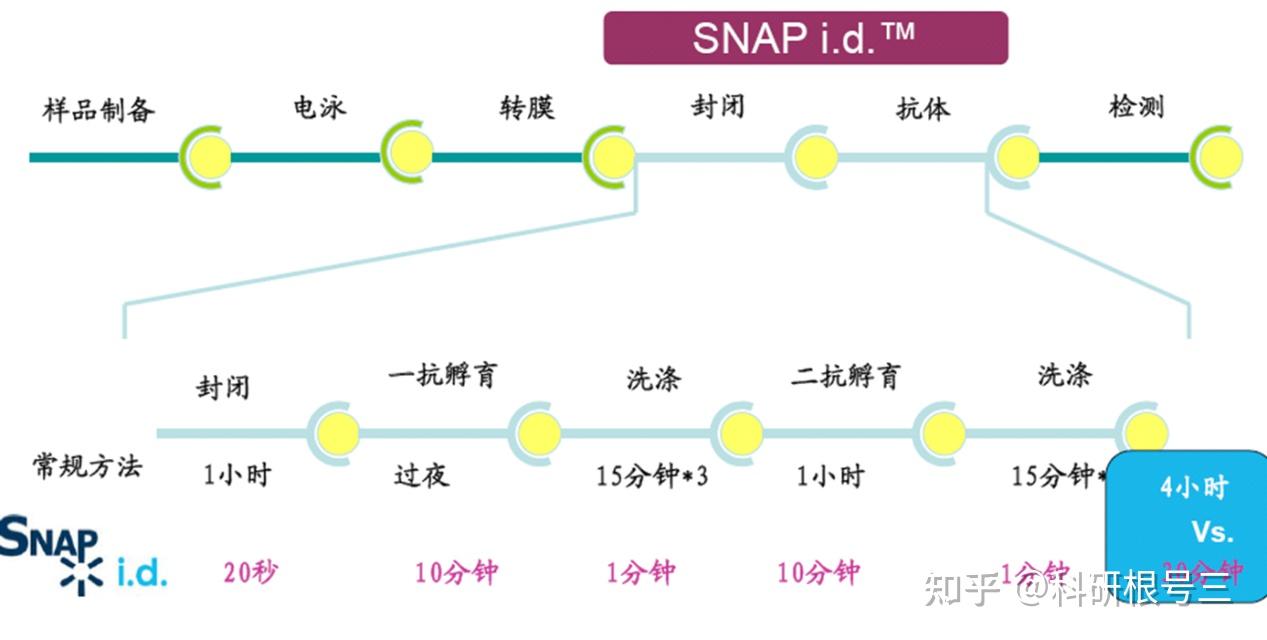
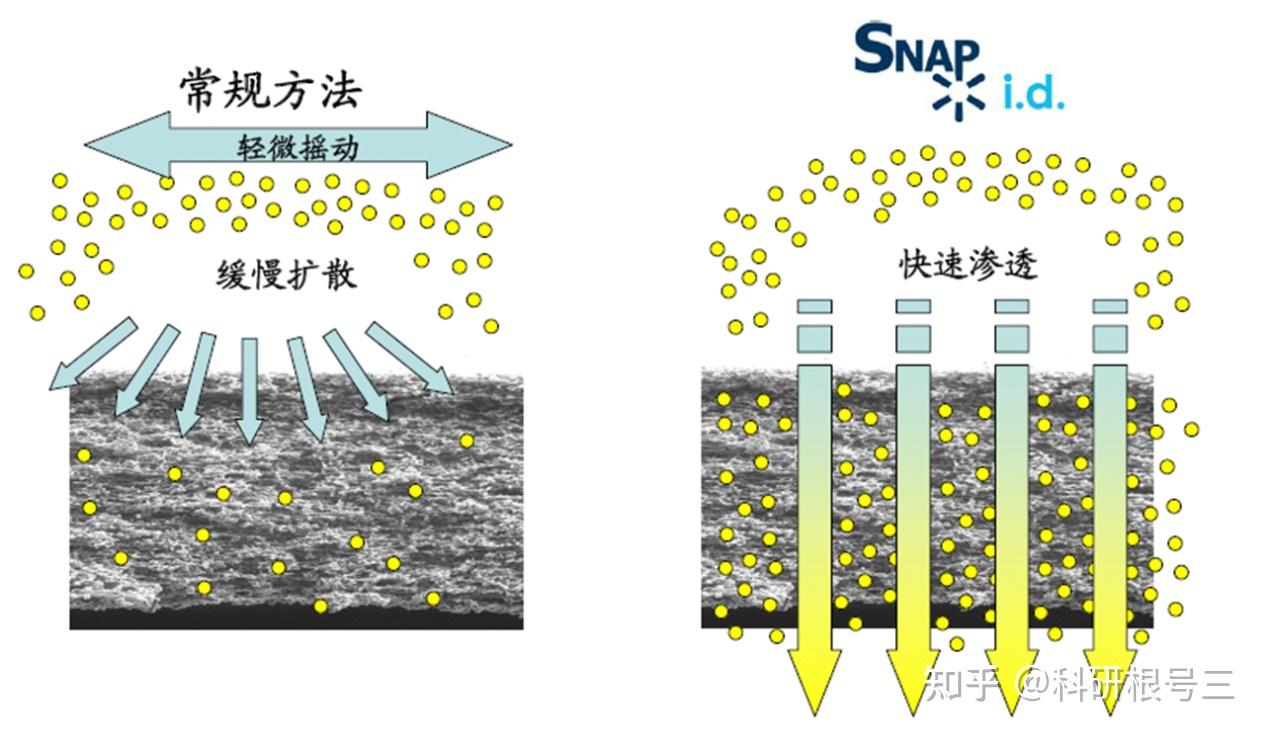
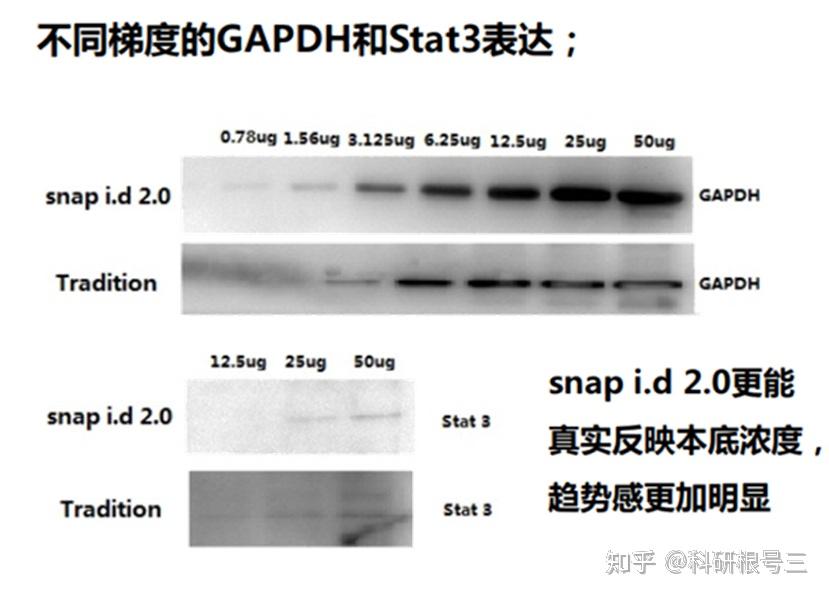
建议使用TBST进行抗体稀释和洗涤。洗涤三次,每次5-10分钟。 对于图中所有抗体,TBST比PBST可以产生更强的信号。
产品推荐:Mouse Anti-rabbit IgG (Light-Chain Specific) (L57A3) mAb #3677; 产品推荐:Mouse Anti-rabbit IgG (Conformation Specific) (L27A9) mAb #3678; 产品推荐:Mouse Anti-rabbit IgG (Conformation Specific) (L27A9) mAb (HRP Conjugate) #5127
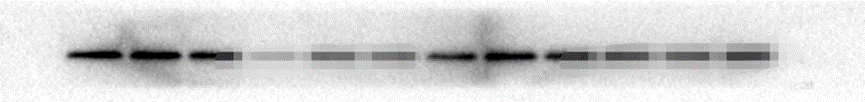
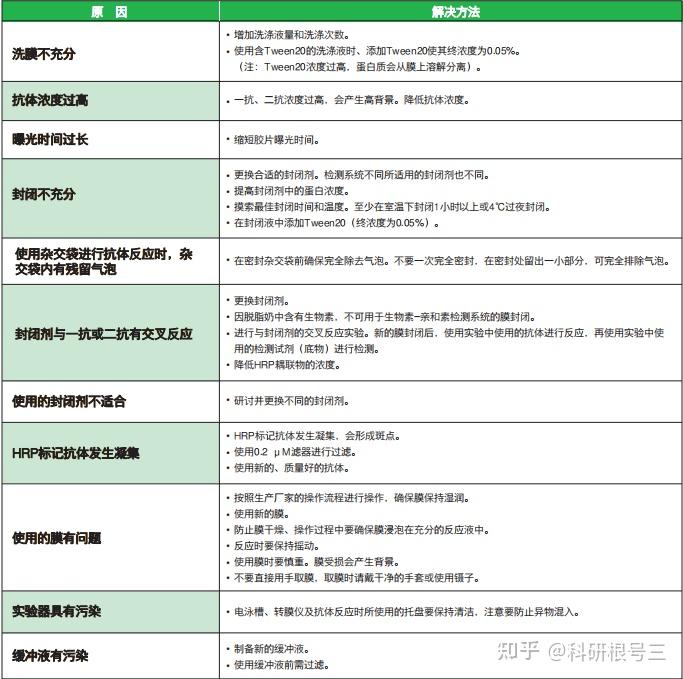
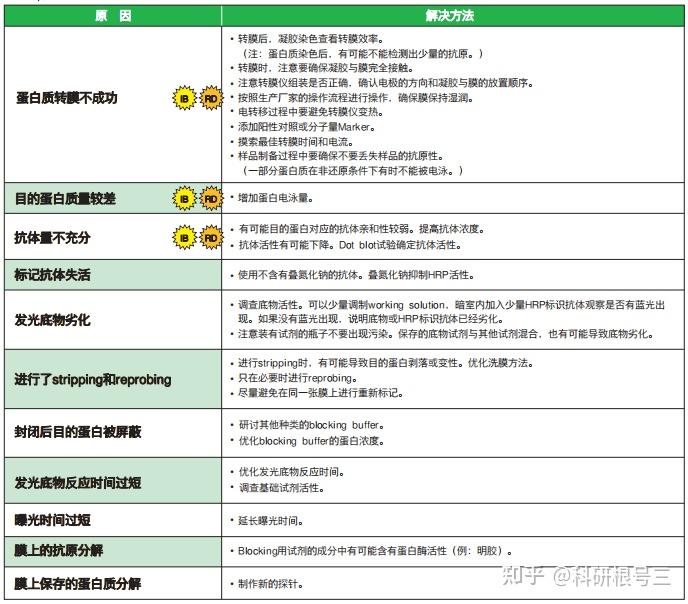
原因:比较多,如果单纯一张没有任何显色的X光片,最可能是一抗加成其他抗体,或者二抗种属加错了,比如兔的加成鼠的。 解决办法:仔细检查抗体是否加错,确认转膜没有问题。 经验:上面的图片展示的是一点信号都没有,如果是这样大部分情况是抗体加错了。如果中间出现了细微的条带,可能原因是蛋白上样量太少,一抗浓度过低,ECL发光液失效。另外如果转膜出现了问题,比如膜放反了,自然是一个白片。
原因:封闭不够好,一抗浓度高,洗膜时间和次数不够 解决办法:降低一抗浓度,增加洗膜时间和次数。 经验:高背景可能是WB中最常见出现的问题,目的条带单一清晰,但是其他地方又弥漫性较为均一的背景(比较连续的)。其实只要我们注意操作规范,不偷工减料就很容易避免,洗膜按照规定来5min*5次或者10min*3次,不要改成5min*3次,或者10min*2次。
原因:一抗非特异性与蛋白结合 解决办法:更换一抗 经验:此种情况绝大多数是因为一抗不好,你无法判断那一条是目的条带。如果实在没有更好的抗体,建议采用阴性对照和阳性对照来确定上述哪个条带是目的条带。当然这种情况下有一种很小几率的可能是一抗浓度太高引起的非特异性结合。
原因:电转中膜和胶之间存在气泡。 解决办法:转膜前去掉膜和胶之间的气泡 经验:我们常常将电转液倒入一个盘子里,倒入的液体不能太多也不能太少,最好的高度是与放上第一层滤纸齐平,然后往滤纸上浇点转膜液,把电泳胶用清水清洗下,将电泳胶平铺到滤纸上,仔细检查滤纸与胶之间是否有气泡,可以左右前后观察,不同方向观察之后确认无气泡,然后再往胶上面浇点电转液,用两只手的拇指和食指轻轻夹住PVDF膜的两侧中间,使膜成U型,然后将U型的底部接触胶的中间,慢慢往两边放下膜,这样一般气泡很少。然后上层滤纸同样用U型的放置方法,用玻璃棒稍微贴实下,然后盖上海绵。注意不要来回赶气泡,这样反而会带入气泡。
原因:中心部位高浓度HRP把底物消耗过快,中间部位底物消耗结束之后就不发光了 解决办法:降低蛋白量,降低一抗和二抗的浓度。 经验:如果你足够迅速,可以在中间部位底物消耗之前就把X光片给定影出来,但是时间很难把握,建议还是从降低蛋白量,降低一抗二抗浓度入手。
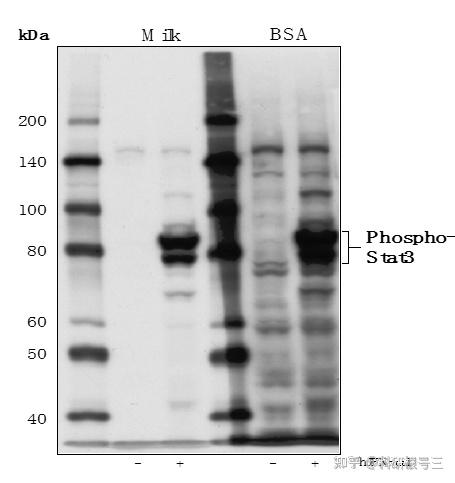
原因:膜上其他部位与一抗或者二抗非特异性结合 解决办法:封闭牛奶一定要纯,封闭结束之后要洗 经验:我们常常是等到要封闭的时候发现没有牛奶,然后匆匆忙忙用PBST/TBST配置,配的匆忙的时候往往不管是否已经完全溶解,如果没有完全溶解的情况下加牛奶倒膜上,会导致很多不溶性颗粒附着在膜上,这就会导致发光时候膜上的黑点。所以牛奶溶解之后,最好静止一下,然后轻轻地吸取上层牛奶进行封闭,封闭结束之后一定要洗三遍之后再加一抗。
原因:蛋白量太大,一抗浓度和时间太长 解决办法:根据情况调整蛋白量,同时一抗浓度和时间也可以缩短。 经验:这种情况很容易出现,因为很多原因都可能导致这一个结果。一般来说,蛋白量都是我们经过很多次摸索得出的最适蛋白量,因此不太可能是蛋白量过多引起的。最有可能的是因为一抗浓度太高,作用时间太长引起的。另外洗一抗和洗二抗千万不要偷工减漏,建议5min*5次,不要但是洗这么多次就把抗体和蛋白洗掉了,你又不是拿刀在上面刮,真正的抗原抗体的结合是通过这种方式洗不掉的。
原因:荧光强度比较高,在压片时,放好之后不小心又轻微移动了一段距离 解决办法:X光片放上去之后,就不要动了,即使放歪了也没关系。 经验:有的时候出现重新刚好在上下位置,并且重影会相对弱一些,不要误以为抗体识别了该蛋白的另外一种异构形式。
原因:膜可能曾经干过 解决办法:在每一步的操作过程中,都需要注意不要让膜干 经验:在封闭的时候,洗一抗,洗二抗,以及发光的时候都时刻需要注意蛋白面不要风干,风干之后结果很可能就是这个样子。注意与高背景区别。
原因:SDSPAGE胶中存在气泡或者某不溶性颗粒 解决办法:配胶过程中要小心,使用无杂质的液体。 经验:很多实验室中使用的不是最新的设备,比如配胶用的海绵垫,如果用了很多年之后,会从下面往上面漏小气泡,当气泡足够小并且胶快凝固的时候,走到中间的小气泡就停留在胶内,并会影响到后面的跑胶。另外配胶用的水,SDS,Tris缓冲液要注意不要有杂质。
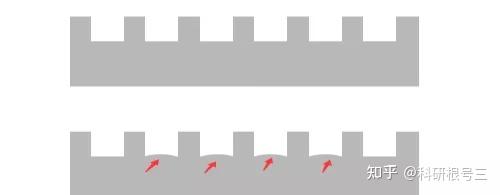
原因:配置胶有问题 解决办法:把胶配好,不合格的胶坚决不用 经验:出现哑铃最大的可能是胶没有配置好,胶凝固后不均一,不知道大家有没有出现如下的配胶情况,下图中示意拔完梳子之后的结果,如果你拔完梳子之后出现图中下面部分的样子,多半会出现哑铃状。另外还有一种可能是样品中含有太多杂质,没有离心下来,然后杂质沉积在孔的中间,蛋白自然被推挤到两边。
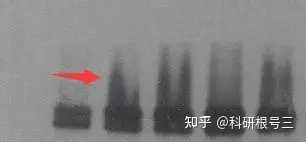
原因:电泳电流不均一 解决办法:换用新的电泳槽; 不使用两边的两孔 经验:一般我们使用的是10孔的的胶,如果你上样刚好10个孔,那么最两头的两个孔肯定会歪曲。另外上样最好在胶的中间,这样电场均一。
本版积分规则 发表回复 回帖后跳转到最后一页
查看 »
微信扫一扫关注本站公众号

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 





 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 雷达卡
雷达卡 发表于 2024-9-9 09:30
发表于 2024-9-9 09:30


 提升卡
提升卡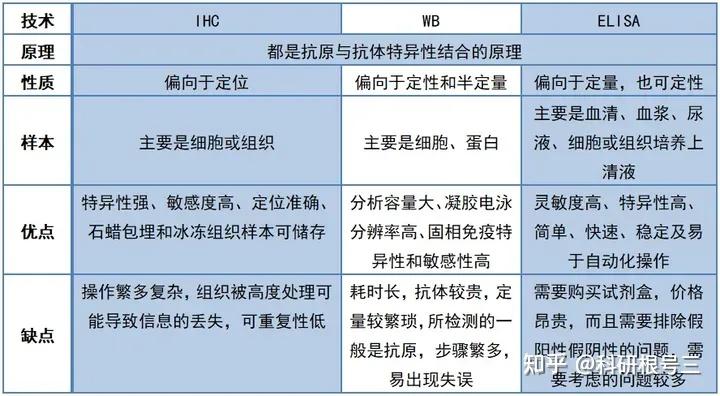






































 发表于 2024-9-9 09:31
发表于 2024-9-9 09:31