不过嘞,这个方法可是费时费力啊,比较几十个还好,比较几百个呢,几千个呢?跑过胶的同学都知道做个电泳有多难,一代测序的每段序列都是靠这么一个个胶板去测的,想想是不是就很恐怖。而且人类基因组有多大呀——3.1个G!3后面跟了9个0,这得测到猴年马月去?所以意义重大的人类基因组计划测了十三年之久。而现在,整个人类基因组的测序大概只需要1天时间。至于成本,当年花费30亿美元的项目,现在华大基因的标价好像只有1300元左右呢。
那这个颠覆性的二代测序技术到底有什么神奇之处[1]?通俗来讲:先疯狂复制一大堆DNA分子,再丧心病狂把长链全打成短短的小片段,然后用非常擅长测小片段的技术,把这些碎末末都测出来(技术细节也暂时略过了,网上非常细致的讲解和演示动画都很多)。问题是这真的靠谱吗?——在有计算机和生物信息学的时候,非常靠谱!让我们回到开头时候提到的这个有点反直觉的结论:使用短读长片段也能对复杂基因组进行测序和从头组装。
这这就是生物信息学中非常基础但也非常重要的一个部分内容:基因组从头组装(de novo assembly)。还是让我们稍微看看最符合直觉的一代测序:由于我们是一小段一小段去测的,那每次我们都和上一次测的有几个碱基的重叠,就可以把它们顺利拼接起来了。那咱现在面对的是满地没头没尾的短片段,可咋办?没关系,让我们擅长大量运算的计算机使用数学魔法!
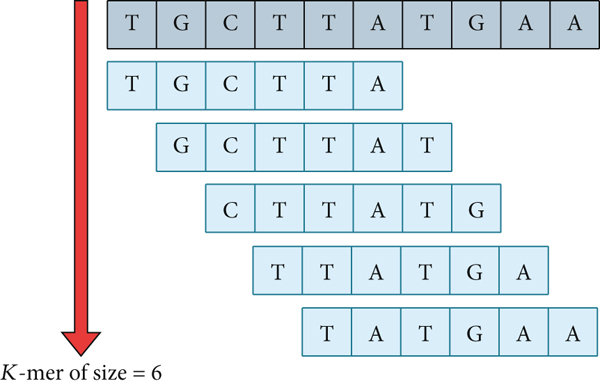
基因组的组装可以用图抽象成一个数学问题[2],图由是一组节点再加上节点之间的边组成的。当我们进行基因组组装时,我们先把已经挺小的片段拆成更小的片段作为节点,由于每个片段的长度为k,所以一般被叫做k-mer,如果在我们拆的时候两个片段是连接着的,就给他们添加一个有向边。没错,我们确实是把已经又多又碎的片段拆得更多更碎了!然而,这样可以让我们通过寻找k-mer之间重叠的方式,来把更多的序列连接在一起,把这些分别连接的小片段“捋”在一起,整合好之后就是完整的序列了。
长度为10的序列被拆成5个6-mer
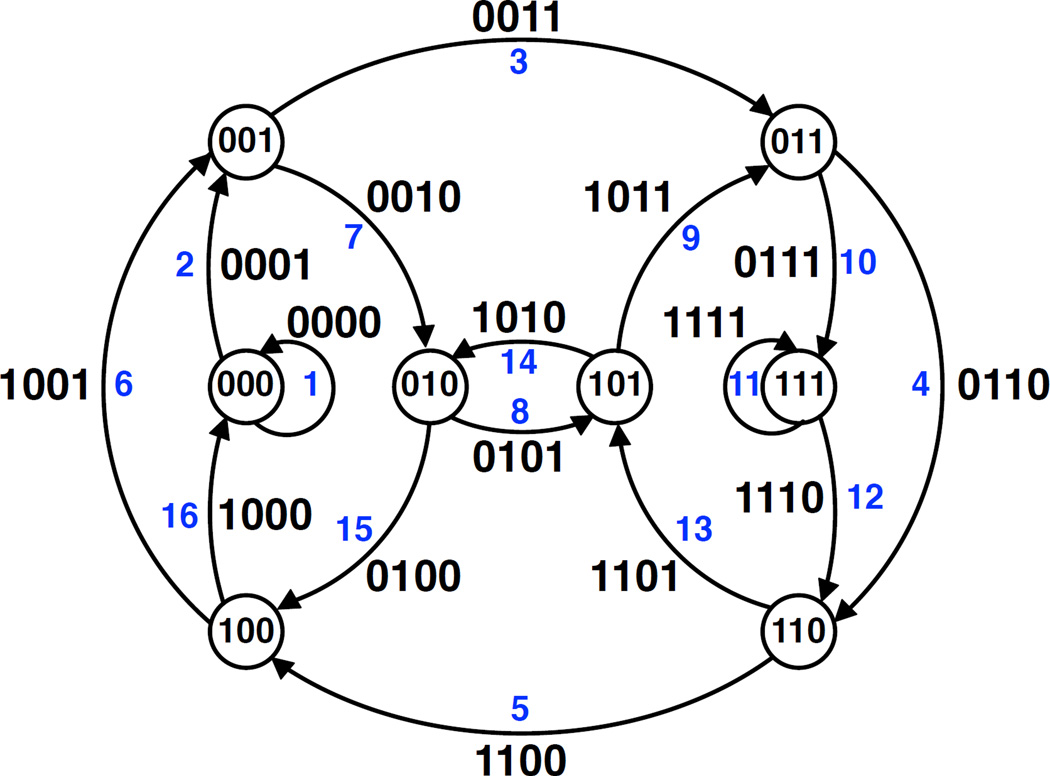
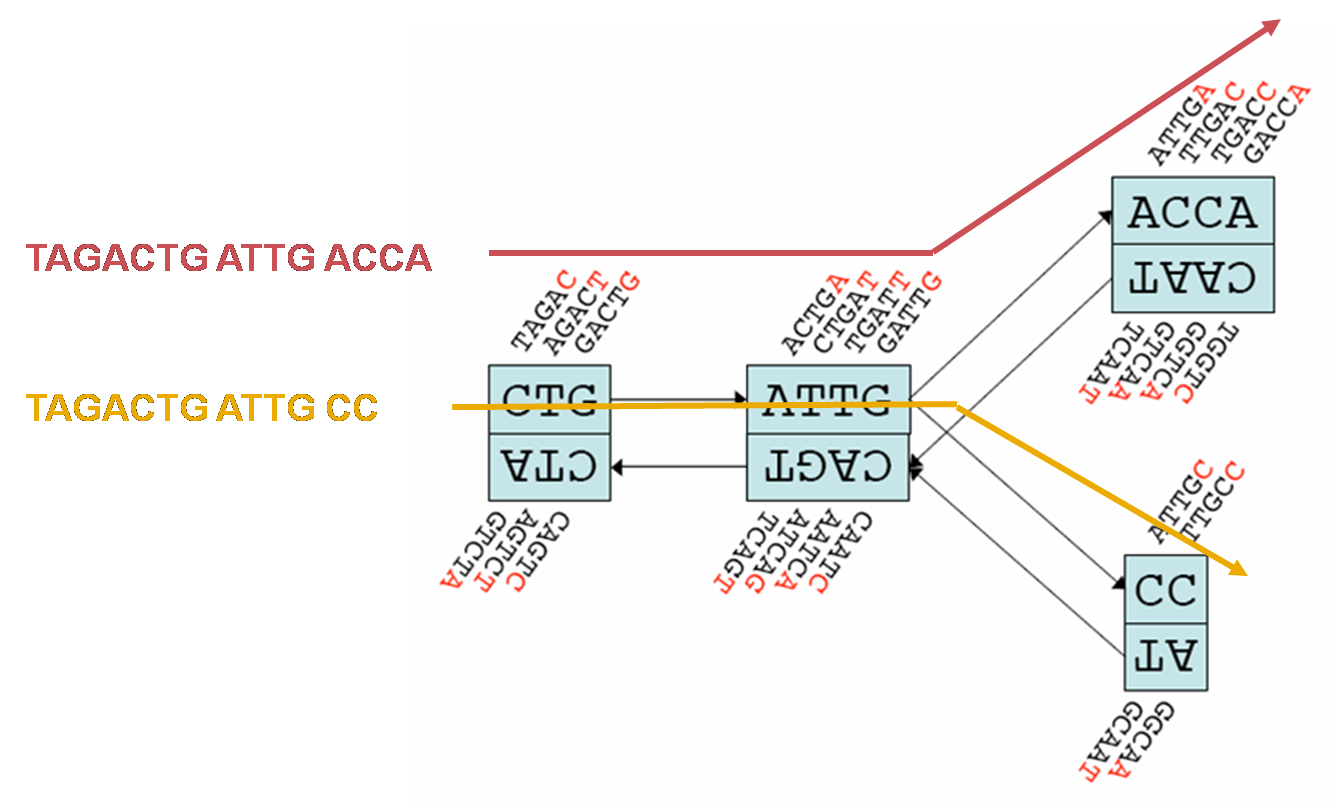
好吧,这好像听起来还是个不小的挑战,没关系,这依然是个图论中经典的数学问题:即找到一个恰好访问图的所有节点一次的循环(称为哈密顿循环问题)。但是如果看做哈密顿循环问题的话,关注点更多是在于节点之间,而在基因组组装中重叠信息可能更为关键。所以我们转向了 de Bruijn 图,通过连接节点来寻找访问图中所有边一次的路径。想象一只蚂蚁想要比访问整个图,比起走过所有节点一次,它现在更想访问每个边一次,感兴趣的小蚂蚁可以去搜搜哥尼斯堡七桥问题和欧拉循环。在计算机的帮助之下,我们的算法可以在有几十亿节点的巨大图中高效地找到欧拉循环,因为对于计算机科学来说,这样的计算是非常轻松而且常用的策略啦。

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 






 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 发表于 2024-9-1 16:46
发表于 2024-9-1 16:46