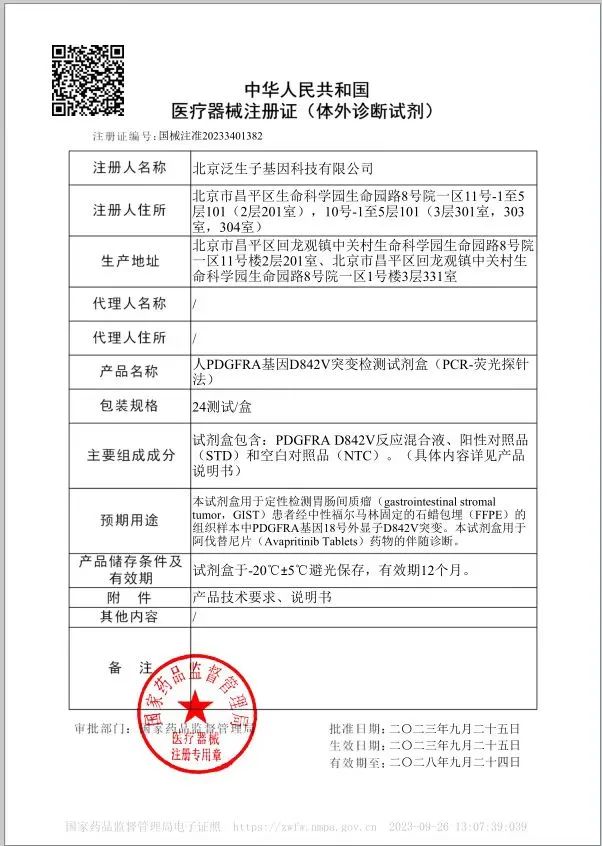
9月27日,基石药业宣布与北京泛生子基因科技有限公司(以下简称“泛生子”)合作开发的泰吉华®(通用名:阿伐替尼)伴随诊断试剂盒“人PDGFRA基因D842V突变检测试剂盒”(以下简称“试剂盒”)获国家药品监督管理局(NMPA)批准,成为中国伴随诊断试剂指导原则出台后NMPA批准的首个以桥接路径合作开发的伴随诊断试剂盒,可通过检测胃肠间质瘤(GIST)患者的PDGFRA基因突变,用于泰吉华®药物的伴随诊断。
据悉,该试剂盒的上市,还实现了多项“首个”,包括:
首个结合中国境内外药效数据批准上市的国产伴随诊断试剂盒;
首个在《抗肿瘤药物的非原研伴随诊断试剂临床试验注册审查指导原则》和《与抗肿瘤药物同步研发的原研伴随诊断试剂临床试验注册审查指导原则》出台后以完整桥接路径上市的伴随试剂盒;
首个药物和伴随诊断均获得NMPA优先审评的案例。
- 2019年底,泛生子和基石宣布合作,该试剂盒仅用三年时间便完成从研发到审批的落地。
- 2021年7月,泛生子宣布,“人PDGFRA基因D842V突变检测试剂盒(PCR-荧光探针法)”获国家药监局优先审批,成为首个进入优先审批程序的国产伴随诊断试剂盒。
PDGFRA基因突变的GIST患者一直缺乏有效治疗药物,而泰吉华®在包含PDGFRA外显子18突变(包括D842V)的不可切除或转移性GIST患者中显示出显著的疗效和可接受的安全性,是中国首个获批用于治疗这一特定患者群体的精准药物。该伴随诊断试剂盒提供了一种合规和准确的筛查方法,用于识别可能从泰吉华®治疗中受益的患者。根据泛生子伴随诊断试剂盒临床试验结果显示,经伴随诊断桥接药物疗效ORR达到92.5%,与临床检测金标准Sanger测序法阳性符合率(PPA)/阴性符合率(NPA)分别达到100%和99.43%,总符合率(OPA)为99.46%。
随着精准免疫疗法越来越普遍,伴随诊断试剂成为必不可少的产品;而纵观全球,伴随诊断试剂的开发晚于药物临床试验开发的情况十分常见;因此,桥接试验成为大部分伴随诊断试剂注册上市的主要路径。2021年12月和2022年6月,国家药监局器械技术审评中心分别出台了《抗肿瘤药物的非原研伴随诊断试剂临床试验注册审查指导原则》和《抗肿瘤药物同步研发的原研伴随诊断试剂临床试验注册审查指导原则》两个伴随诊断试剂相关的审查指导原则。业内普遍认为这一系列政策落地,在规范伴随诊断试剂盒注册要求的同时,也提高了伴随诊断企业的注册门槛。而此次泛生子的产品获批,作为新规实行以来首个以桥接试验方式获批的试剂盒,具备了成功“过河”的经验,为后来者提供了可参照的范本。

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号