前晚,央视315晚会如期而至 大家对于315将爆出何种猛料热情不减 不出所料,热搜又爆了!
虽然今年的315晚会整体略显疲态,没有锋芒毕露的剑指大型企业,甚至像医美产品、水军事件、短信骗局之类话题都是老生常谈,但其通过电视叩问市场良知的初衷却丝毫未变。 315的本质是监督,随着互联网时代的到来,人人都是自媒体,天天都是315将成为一个常态。对于企业来说,社会监督常态化,企业的预防也应常态化。日常做好管理,就是最好的预防。
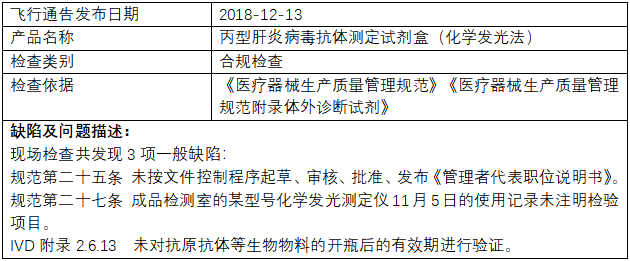
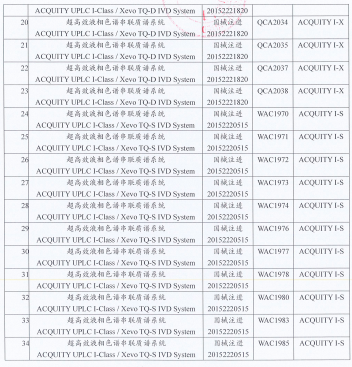
2023年“315”晚会 因此,小编整理了近2年国家药监局飞行检查、医疗器械抽检情况,汇总出IVD中突出的一些生产质量问题进行分享。  (一)国家药监局飞行检查通报: 2022年,国家药监局核查中心组织开展了医疗器械生产企业飞行检查工作,发现23家企业存在一般项目不符合《医疗器械生产质量管理规范》及相关附录要求。 存在问题主要有: 机构与人员方面 1. 企业生产、质量与技术人员等学历、专业及工作经历与公司规定要求不符,培训不到位,未能提供从事影响产品质量工作的人员有针对性的培训记录(如生产负责人、专职检验人员等); 2. 企业负责人及各部门负责人员对最新法律法规不熟悉; 3. 人员从事工作无相关的任命书,人员对从业的相关业务基本知识缺乏 4. 同一人兼任组织机构中独立部门的负责人,具体工作跨部门人员承办; 企业十万级洁净室使用的一次性无菌工作服不含脚套,不能包盖脚部。 厂房与设施方面 1. 物料储存缺乏明显标识或物料的帐、卡、物数量、有效期等信息不一致; 2. 产品紧靠墙壁存放; 3. 洁净区人员数量限定管理制度中未规定缓冲间的洁净区人员数量上限,洁净区房间外未张贴人员上限提示; 4. 湿度与规定不符; 5. 排风过滤器未定期检查和更换,空调过滤网有较多灰尘; 6. 洁具间与洁净走廊间的门向洁具间方向开启; 7. 十万级洁净车间下水管道采用直管,未设置防倒灌装置。下水槽及地漏敞开,有潜在污染的风险。 设备方面 1. 生产及检验设备标识有误或设备状态标识不全; 2. 未按设备说明书要求纳入操作规程; 3. 未按相应文件开展验证或未对设备的再验证周期作出规定; 4. 主要生产设备未提供相应的验证及确认记录; 5. 计量器具的精度、量程与实际使用不相符或检定校准已过有效期; 6. 空气净化系统、烘干机、冷藏、冷冻设备温度不达标,或维护不到位; 7. 灭菌间内相关设备维护不到位或未能及时维修; 8. 温度监测设备在温度超限时报警功能未启动,仓库内温度不达标等 9. 洁净区内部分门不能密闭,相关设施不能及时维修 10.灌装设备存在污染物 文件管理方面 1. 卫生管理相关程序文件缺失 2. 检验项目“有效使用量”涉及称量和密度计算,未记录试验温度和湿度; 3. 企业的《不良事件控制程序》未按要求及时修订; 4. 原始检验记录中所附的试验照片仅保留纸质版未保留电子数据;检验室冰箱存放的试验样品未建立台账; 5. 体系文件与产品技术要求、工艺规程不一致; 6. 文件错误处直接进行修改,未注明修改人和修改时间; 7. 部分文件记录易混淆,不易于识别和检索;重要文件未编号 8. 原材料库中的产品批号与货位示批号不一致 设计开发方面 1. 无法提供完整的产品设计开发文档;未保留原始检测数据,或无法提供证明该检测数据真实的证明材料 2.设计开发更改评审记录不完整或设计更改后,未经过风险分析和评审就直接开始做验证。 3.批量生产前由研发部门根据抗体活性进行抗体使用量的确认,但配对规程未明确配对参考品浓度、抗体浓度筛选范围依据。 生产管理方面 1. 记录不完整(如:缺少规格型号、生产设备、参数、产品批号),或批生产记录与检验记录不一致或无法追溯; 2. 现场检查洁净生产车间,用于操作人员裸手消毒的酒精容器未标示配制日期和失效日期,不符合《洁净室(区)管理制度》要求标明配制日期和失效日期的规定; 3. 抽查某批生产记录,未记录设备名称、设备编号、工艺参数等信息; 4. 未按要求及时进行现场生产记录、未将洁净区内使用的消毒剂更换周期、频次、效期等内容进行验证确认; 5. 新购置的设备未经设备验证就投入使用或相关设备清场不彻底; 6. 未规定防护要求; 7. 现场查看配料环节生产环境,无粉尘防护的措施; 8. 10万级包装间内发现周转箱敞口叠放。 质量控制方面 1.主要原材料的检验项目未记录实测数值,只有结论; 2.未按文件规定进行留样观察及记录; 3.实验室立式灭菌器、手提式压力蒸汽灭菌器的压力表标识在校准期限内,安全阀未见检定标识; 4.部分仪器未能按标准进行校准; 5.未明确中间产品存放时间,未明确长期储存中间产品使用前的质控要求; 6.产品初始污染菌检测记录中没有给出修正系数值,直接给出检验结果; 7.洁净区风淋室环境未进行定期监测; 8.企业成品检验规程规定阳性符合率为检测的判定标准为检测2份阳性参考品,批检验记录与成品检验规程一致,但成品检验报告中阳性符合率的判定标准为检测10份阳性参考品。 9.过程检验规程缺少滴斗和瓶塞穿刺器粘接密封性的检验要求。 不良事件监测、分析和改进方面 1.企业对收集到不良事件信息建立了清单,但未能对产品潜在不合格及其原因进行有效汇总、分析和确定; 2.企业采取纠正措施,更改产品检验规程的最低检出量项目的半成品检验要求,但成品检验要求未做相应改变; 3.企业按不良事件监测和再评价控制程序开展两轮临床试验受试者长期随访研究,但随访研究未制订统一方案和工作内容要求; 4.未对产品检验过程中物料发生溢胶、气密性不合格等问题进行数据识别、分析和记录; 5.企业对产品进行了退换货原因分析,但未能对产品退货原因进行与质量有关的分析; 其他 - 药监局官方网站上的飞行检查: (二)国家医疗器械抽检: 2021年12月22日,国家药监局发布国家医疗器械监督抽检结果的通告(第5号)(2021年第105号),涉及18个品种,共39批(台)产品不符合标准规定。 其中,和IVD行业相关的产品有: (一)γ-谷氨酰基转移酶测定试剂(盒)1批次:涉及线性区间不符合标准规定。 (十二)手术衣2批:涉及胀破强力-干态(产品关键区域)、胀破强力-干态(产品非关键区域)、胀破强力-湿态(产品关键区域)不符合标准规定。 (十三)四氢大麻酚酸检测试剂盒(胶体金法)2批:涉及阳性参考品符合率、最低检测限、重复性、物理性状不符合标准规定。 (十七)一次性使用无菌手术膜2批:涉及水蒸气透过性不符合标准规定。 (十八)医用超声雾化器1台:涉及设备或设备部件的外部标记、指示灯和按钮不符合标准规定。 (三)主动召回: 部分有社会责任感的企业,在发现缺陷产品后,坦荡面对社会监督,主动向主管部门报告,及时召回产品。 1. 全自动荧光PCR分析仪cobas z 480主动召回 ● 召回原因:某企业总部收到cobas 4800 IVD测试中假阳性或结果无效的投诉,调查发现,全流程测试和PCR流程测试受该问题影响,UDF(客户自定义软件)测试流程不受该事件影响。某企业对其生产的全自动荧光PCR分析仪cobas z 480(注册证号:国械注进20163222584)主动召回。 2. 超高效液相色谱串联质谱系统)主动召回 ● 召回原因:产品超高效液相色谱串联质谱系统(型号: ACQUITY I-S)和超高效液相色谱串联质谱系统(型号: ACQUITY I-X)缺少系统标签决定对涉及产品进行主动召回。 ● 召回产品:某企业对超高效液相色谱串联质谱系统 ACQUITY UPLC I-Class IVD/Xevo TQ-D IVD System(注册证号:国械注进20152221820,型号: ACQUITY I-X)和ACQUITY UPLC I-Class IVD/Xevo TQ-S IVD System(注册证号:国械注进20152220515,型号: ACQUITY I-S)进行主动召回,此次召回不涉及实物召回,召回级别为三级。
小编有感: 目前,国内IVD企业超过2000家,产品技术同质化突出;在重度内卷之下,众多企业将目光聚集到了海外市场。虽然海阔凭鱼跃,但要想推动中国的IVD行业真正走向世界,就要实现从制造到质造再到智造的转型升级。在这之前,修好”内功”,从产品出发,严把质量关,才能为赢得全球市场打下坚实基础。 文章内容由“MIR医学仪器与试剂”编辑整理,转载请注明来源。 |


 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号