在日常工作中,小编发现很多人都分不清PCR、qPCR、RT-PCR、Hot Start PCR、Nested PCR……今天我们就来了解一下PCR技术的基础原理,以及目前常用的PCR技术。 什么是PCR? PCR又称为聚合酶链式反应。是一种用于放大扩增特定DNA片段的技术,其能让NDA片段数目以指数型增长。在PCR技术出现之前,人们针对DNA进行研究的时候,通常都要十分的小心地使用这些核酸样品,因为在体外扩增的PCR技术出来之前,每份DNA样品都十分珍贵,研究者只能节约的使用。而PCR技术的出现打破了这种限制。其原理与体内DNA的复制类似,可看作是生物体外的特殊DNA复制,无论是在任何地方只要我们能分离出一丁点的DNA,我们就能用PCR技术进行放大。 PCR过程大致分3部 变性—退火—延伸 1 变性:(90℃-96℃):双链DNA模板在热作用下,氢键断裂,形成单链DNA。 2 退火:(60℃-65℃):系统温度降低,引物与DNA模板结合,形成局部双链。 3 延伸:(70℃-75℃):在Taq酶(在72℃左右,活性最佳)的作用下,以dNTP为原料,从引物的3′端开始以从5′→3′端的方向延伸,合成与模板互补的DNA链。 如下图所示,在经历一次变性-退火-延伸的流程之后,原本只有1条的DNA双链变成了2条;2次之后是4条;以此类推,只要经历几十个循环数,就能产生大量的相同DNA片段。
PCR实验流程示意 以上就是常规PCR的正常流程。那在扩增完成之后我们需要对扩增出来的DNA进行检测,常规的PCR技术通常只能借助凝胶电泳手段。这里不对电泳技术做过多介绍,其是根据DNA的分子量大小不同对DNA进行分离,从而进行鉴定以及纯化DNA的技术。借助凝胶电泳可以判断扩增出来的DNA片段条带大小是否与目的条带大小一致,从而知道是否得到了想要的产物;看是否有杂带,判断扩增是否具有特异性;看是否有引物二聚体判断引物的设计好坏等等。 关于qPCR和Real-time PCR 很多人误以为RT-PCR是Real-time PCR的缩写,因此将二者混为一谈,但实际上是不一样的。Real-time PCR又叫做实时荧光定量PCR,也就是大家所熟知的qPCR,所以这二者才应当是同一个东西。在刚才介绍PCR后续检测方法的时候,我们说了通常只能借助凝胶电泳去分析产物,但实时荧光定量PCR借助荧光化学物质让我们在PCR的过程当中就可以检测到DNA的扩增情况,从而不需要再去跑电泳了,极大的简化了操作者的实验步骤。 qPCR的实验原理 简单来讲,qPCR就是在整个扩增的过程中,引入了荧光基团,随着DNA数量的成倍增加,反应体系中的荧光信号也会增强。借助对荧光信号的强弱进行检测,从而对DNA扩增的情况进行定量分析。 目前根据不同的荧光物质,我们可以大致将qPCR分为两种。一种是用荧光染料,主要为SYBRGreenⅠ染料,该染料能与DNA双链结合,结合之后会显示荧光信号,而在没有结合的时候几乎检测不到荧光信号,随着拷贝数的增加,荧光信号增强。而还有一种是使用荧光探针,又叫TaqMan探针法,其原理是根据目的基因设计一个能与之特异性结合的荧光探针,该探针包含两个基团:一个荧光基团和一个淬灭基团,正常情况下因为淬灭基团的存在导至荧光基团无法发出荧光,而在扩增时探针结合到DNA的模板上,并且扩增到探针位置时两个基团被切开,从而荧光基团可以发出荧光,同样随着拷贝数的增加,荧光信号增强。
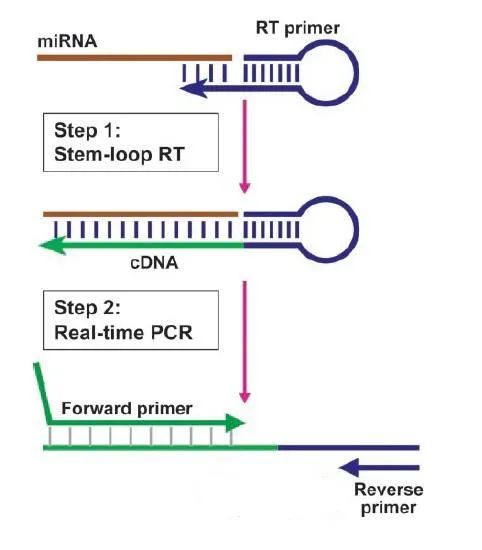
TaqMan探针法原理图 借助这种定量关系,人们可以绘制出PCR的扩增曲线,该曲线是荧光信号随着循环数变化而变化的曲线,借助该曲线就可以对PCR的情况进行分析,理想的曲线应该是呈现“S”型的增长,但是关于曲线分析的内容比较复杂,在这里暂时不做过多赘述。至少我们知道借助该技术我们可以不用在每次做完PCR之后去辛苦的跑电泳,也可以更精确的分析我们的PCR结果。因此该技术被越来越广泛的应用。 值得一提的是,在疫情当下,我们在做完核酸采样之后到底如何去判定被检者的阴性还是阳性,使用的便是qPCR技术,倘若在经过扩增一定循环数之后能检测到荧光信号,我们可认为被检者为阳性,反之则阴。 RT-PCR RT-PCR相较于前者来说就简单得多。其全称为反转录PCR,或逆转录PCR。首先我们要清楚什么是反转录,我们一般将遗传信息从DNA流向RNA的过程称为转录,反之,以RNA为模板合成DNA的过程我们称为反转录。 该技术的一般过程为,首先以RNA为模板在反转录酶的作用下合成其对应cDNA,再以cDNA为模板进行普通的PCR过程。归根结底,其就是在PCR过程前加了一个反转录过程,那在这之后我们既可以用普通PCR手段,也可以采用qPCR进行扩增。比如病毒通常是以RNA为遗传物质,我们在进行病原体检测的时候只能提取到它的RNA,所以是需要进行反转录的。
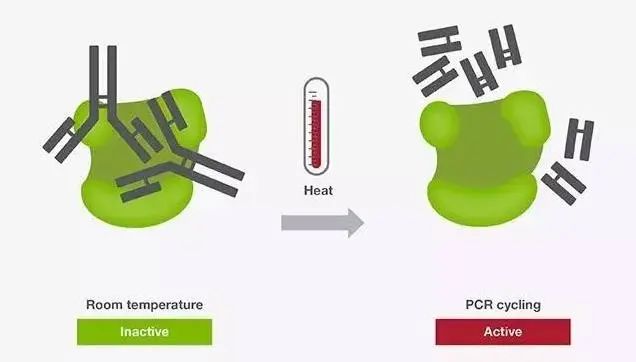
热启动PCR(Hot Start PCR) 热启动PCR常用于增强PCR扩增的特异性。热启动PCR主要借助一种酶的修饰物(如抗体、亲和配体、适体或化学修饰物)来抑制常温下DNA聚合酶的活性。这种修饰使得在PCR反应体系配制阶段引物与模板、引物与引物之间的结合能力降低,从而避免了非特异性扩增。由于DNA聚合酶在常温下的活性被抑制,热启动技术为在常温下配制多个PCR反应体系提供了极大的便利,而无需牺牲PCR反应的特异性。
基于抗体热启动技术的DNA聚合酶 当反应体系配制好后,在反应初始加热步骤,酶修饰物在高温下(通常高于90 ℃)被释放,使得DNA聚合酶被激活。激活时间和温度根据DNA聚合酶及热启动修饰物的不同而有所差别。对于某些聚合酶,激活和预变性合并成了一步。 降落PCR(Touchdown PCR) 另一种提升PCR反应特异性的方法是调整PCR循环的参数。在降落PCR中,前几个循环的退火温度设定为比引物的最高退火温度(Tm)再高几度。较高的温度有助于避免产生引物二聚体及非特异性引物与模板形成的复合物,因此可以减少不希望出现的扩增。就这一点来说,较高的退火温度可减少非特异性PCR产物,在PCR起始阶段促进特异性的扩增。
降落PCR 虽然较高的退火温度可阻止引物二聚体形成和非特异性引物结合,但同时较高的退火温度会加剧引物与目标序列的分离,导至PCR的产量降低。为了克服这个问题,在开始的几个循环中,退火温度通常设置为每个循环降低1℃,以获得足量的预期扩增子。当退火温度降低到最佳温度时(通常比最低的引物Tm低3-5℃),剩余的循环都维持此退火温度。通过这种方法,在PCR过程中,所期望的PCR产物得到有选择性的增加,而几乎不会出现非特异性的产物。 通过以较高的温度起始,再随着循环逐渐降低到最佳退火温度(黑色曲线),这种PCR方法可提升反应特异性(黄色曲线)。目标扩增子的产量(绿色曲线)相应的得到富集。 巢式PCR(Nested PCR) 巢式PCR是常规PCR的一种演变,其增强了反应特异性和目标扩增子的产量。在此方法中,需要设计两对引物:一对(外引物)在目标扩增区域的侧翼,而另一对引物(巢式引物)对应于所扩增DNA的准确区域。第一轮PCR的产物之后作为第二轮PCR的模板。
巢式PCR 如果第一对引物(外引物)由于引物错配扩增出了非特异性产物,第二次能在错误片段上进行引物配对并扩增的概率极低,所以通过第二对引物的扩增,PCR的特异性得到了提升。进行两轮PCR的另一个好处是这种方法有助于从有限的起始DNA中扩增得到足量的产物。 多重PCR(Multiplex PCR) 多重PCR可实现在同一反应管中同时扩增多个不同的片段。多重PCR不仅意味着节约时间、试剂和样本,同时使得多个扩增子进行同时比较成为可能。
单个PCR与多重PCR的比较 在单个PCR中,每个反应含有一个引物对来扩增一个目的片段。在多重PCR中,在同一个反应中使用多个引物对来扩增多个目的片段。 当一个反应管中存在多个引物对时,如在多重PCR中,非特异性扩增和扩增效率的降低是最关注的问题,因为反应的优化不可能针对所有的引物对和片段。因此,引物的设计至关重要,以减少错配引起的非特异性扩增。引物序列对于目标片段应该是独特的,且所有引物的Tm值差异应在5℃内。在进行多重PCR前,每个引物对应在单个反应中验证其特异性和扩增效率。而且,不同大小的扩增子应在凝胶电泳中可区分开,以便鉴定。除了引物设计和扩增子大小,热启动DNA聚合酶和特殊优化的PCR缓冲液对于多重PCR也是必需的,它们可有助于扩增的成功率和扩增的特异性。 虽然多重PCR通常用于终点法PCR,但由于其在多重标记和检测中的能力,将其用于实时定量PCR也变得越来越流行。多重实时定量PCR也常用于遗传标志物的检测,用于身份鉴定。 其它的PCR技术 其实除了以上介绍的几种PCR之外,现在还有快速PCR(Fast PCR),以及快速PCR(Fast PCR)、直接PCR(Direct PCR)、高GC含量PCR(GC-rich PCR)、长片段PCR(Long PCR)、反向PCR(Inverse PCR)、定量PCR(Quantitative PCR)等等。但其本质都离不开最开始说的PCR的三个步骤,虽然PCR的基础概念仍然没变,但新的PCR方法提高了PCR扩增的结果,一直在推动和简化分子生物学研究。 资料来源:中国科学院大学、网络,由MIR医学仪器与试剂整理,转载请注明来源。 |

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号