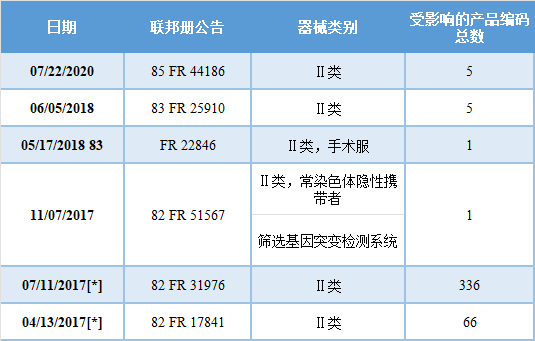
大多数I类和II类器械都被豁免于上市前通知[510(k)]要求。它们也可能被豁免于现行的器械GMP(GMP),以及质量体系(QS)规定的要求。 免于510(k)要求的I类或II类器械仍然必须遵守其他要求(称为监管控制),除非该器械在该器械类型的法规中明确豁免于这些要求。 要确定一个器械是否被豁免于510(k)或GMP要求,请搜索FDA的产品分类数据库(https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPCD/classification.cfm)。 510(k)豁免 大多数I类和一些II类器械在某些限制条件下可免于510(k)要求(见《联邦食品、药品和化妆品法》第510(l)(1)条)。如果FDA确定不需要510(k)来为器械提供合理的安全和有效性保证,则该器械可以免于510(k)要求(见Federal Food, Drug, and Cosmetic (FD&C) Act第510(l)和510(m)条)。可能不受510(k)要求约束的器械包括: ➤ 修正前器械,这部分内容请参考FDA的指南“Intent to Exempt Certain Unclassified Medical Devices from Premarket Notification Requirements”(https://www.fda.gov/regulatory-information/search-fda-guidance-documents/intent-exempt-certain-unclassified-medical-devices-premarket-notification-requirements); ➤ 由FDA特别豁免的第一类器械或根据第513条归类为第一类的器械(有某些例外); ➤ 由FDA特别豁免的II类器械。 术语“修正前器械”是指在1976年5月28日颁布Medical Device Amendments之前在美国合法销售的器械,并且没有: ➤ 自那时起有重大改变或修改; ➤ FDA没有确定需要进行上市前批准(PMA)申请,以合理保证该器械的安全性和有效性。 如果I类器械是用于防止健康损害的重要用途,或呈现潜在的不合理的疾病或伤害风险,则不能豁免510(k)通知要求。 免于510(k)要求的I类和II类器械的清单可在医疗器械豁免510(k)和GMP要求网站上找到(https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpcd/315.cfm)。 对510(k)的上市前通知要求的豁免只适用于那些在该通用类型中具有现有的或可合理预见的商业分销器械特征的器械。 通用类型的I类或II类器械的豁免的一般限制规定在每个器械分类条例中(21 CFR 862.9至892.9)。因此,当器械符合第862.9至892.9节所述的任何条件时,豁免器械的制造商仍需在引入器械或交付器械以引入商业分销之前提交上市前通知。 此外,FDA可能部分限制510(k)要求的豁免,使其适用于分类法规中的特定器械,确认一个器械的510(k)豁免状态和任何可能适用的限制是很重要的。 其他有用的资源包括21 CFR 862-892,产品分类数据库和FDA之前在联邦公报上公布的豁免公告(见下表的例子)。FDA器械和放射健康中心的工业和消费者教育部门(DICE)也可以帮助你确定你的器械的适当要求。 Cures Act豁免 21st Century Cures Act第3054条修订了FD&C Act第510(l)和510(m)条。经修订后,这些条款要求FDA在一定的时间范围内,通过在联邦公报上公布,分别确定任何类型的I类或II类器械,FDA确定不再需要根据FD&C法案第510(k)条提供报告以提供合理的安全和有效性保证。 下表列出了FDA在Cures Act通过后根据这些法定要求采取的行动,这不包括因申请豁免上市前通知要求而实施的豁免。 要确定受影响的具体产品代码,请在FDA网站搜索联邦注册公告(Federal Register Notice)。
[*]这些通知包括FDA根据21st Century Cures Act规定的程序确定的不再需要上市前通知的产品代码清单。在12月30日。2019年,FDA发布了一项最终命令(84 FR 71794),以修改相关的编纂语言,以反映这些豁免。 质量体系条例/GMP豁免 所有医疗器械都要遵守质量体系条例(21 CFR 820),包括“现行GMP”或“GMP”,除非21 CFR 820中指出有例外或豁免。无论哪种类别,你都应该参考器械的具体分类规定,并确认监管要求。 |

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号