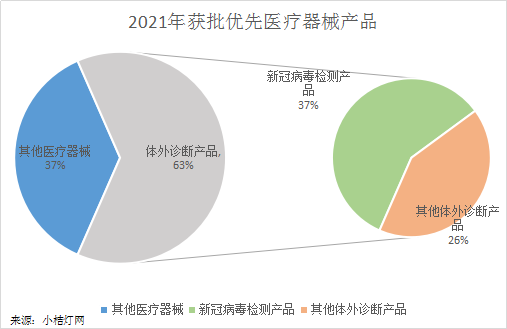
为了满足人民群众对医疗器械的临床需求,我国先后出台了一系列扶持政策鼓励产业发展,如《健康中国2030规划纲要》、国家科技重大专项或重点研发计划、《创新医疗器械特别审批程序》、《医疗器械优先审批程序》等。 所谓“优先审批程序”就是指根据申请人的请求,对纳入优先审批程序的医疗器械产品,在注册申请前及审评审批过程中,设立特别通道,优先进行服务的程序,能够大幅缩短产品上市时间。 《医疗器械优先审批程序》规定,对符合下列条件之一的境内第三类和进口第二类、第三类医疗器械注册申请,实施优先审批: (一)符合下列情形之一的医疗器械: 1.诊断或者治疗罕见病,且具有明显临床优势; 2.诊断或者治疗恶性肿瘤,且具有明显临床优势; 3.诊断或者治疗老年人特有和多发疾病,且目前尚无有效诊断或者治疗手段; 4.专用于儿童,且具有明显临床优势; 5.临床急需,且在我国尚无同品种产品获准注册的医疗器械。 (二)列入国家科技重大专项或者国家重点研发计划的医疗器械。 (三)其他应当优先审批的医疗器械。 根据国家药监局2021年度医疗器械注册工作报告显示,2021年获批优先医疗器械产品19项,其中体外诊断产品12项,占总数的63.16%,其中新冠病毒检测试剂产品数量又占体外诊断产品的58.33%。
新冠病毒检测试剂产品占优先审批医疗器械数量的37% 2021年共批准注册新冠核酸检测试剂4个,分别来自迪安生物、中元汇吉、凯普生物和思路迪生物,其中思路迪生物的是新冠+甲流+乙流联检试剂;批准注册新冠抗体试剂3个,分别来自艾维可生物、康华生物和新产业;另外批准注册了一个全自动医用PCR分析系统,来自致善生物。 新冠疫情的影响想必不用多说,每个人都深有体会。据统计,新冠疫情导至全球超过580万人死亡,超过4.18亿人感染,80%以上劳动人口受到影响,几十亿人被隔离,给全世界人民造成了不可磨灭的生理、心理和经济三重创伤。根据联合国的评估,因冠疫情带来的经济损失总值,已经超越最近100年最大的危机。 2021年批准注册了两项乙肝病毒检测试剂 系上海仁度的乙型肝炎病毒核酸测定试剂盒(RNA捕获探针法)和苏州天隆的乙型肝炎病毒(HBV)核酸测定试剂盒(PCR-荧光探针法)。 乙型肝炎病毒( Hepatitis B Virus,HBV)是一种球形DNA病毒,只对人和猩猩有易感性,引发乙型病毒性肝炎疾病,其中部分慢性肝炎可演变成肝硬化或肝癌。据国家卫健委报告显示,我国40种法定传染病中,病毒性肝炎是发病人数最多的传染病。乙肝病毒感染者约9000万例,其中,慢性乙型肝炎患者约2000万-3000万例,且逐渐呈老龄化趋势。
据世界卫生组织发布的《2016-2021年全球卫生部门病毒性肝炎战略》统计,全球急性感染以及与肝炎相关的肝癌和肝硬化,每年造成140万例死亡。据国家癌症中心的数据显示,我国每年新发的肝癌人数高达37万,其中90%患有乙肝,每年死亡约32.6万,居恶性肿瘤的第二位,并呈现逐年递增的趋势,乙肝已成为世界范围内严重危害人类健康的疾病,治疗工作形势严峻。 乙肝五项的检测方法主要有胶体金法、酶联免疫法(ELISA)、荧光免疫法、化学发光法和核酸检测等。HBV-DNA是乙肝病毒存在直接的依据,是评价乙肝病毒复制水平、传染性强弱、药物疗效的指标,可检测出隐匿性慢性乙型肝炎。乙肝核酸检测具有明显的特异性、灵敏性优势。 2021年批准注册1项呼吸道病原菌检测试剂 系百康芯生物的呼吸道病原菌核酸检测试剂盒(恒温扩增芯片法)。该产品用于定性检测有呼吸道症状的患者痰液中耐甲氧西林葡萄球菌(mecA 基因)、金黄色葡萄球菌、大肠埃希氏菌、肺炎克雷伯菌、铜绿假单胞菌、鲍曼不动杆菌、嗜麦芽窄食单胞菌等7种临床常见下呼吸道病原菌及耐药基因。采用恒温扩增技术,利用荧光染料掺入法进行实时荧光检测,阳性样本扩增检测时会产生“S”形扩增曲线,一步完成对靶基因的扩增和检测。 呼吸道感染是由致病微生物入侵呼吸道并进行繁殖所致的疾病,可分为上呼吸道与下呼吸道感染。据世卫组织报告,下呼吸道感染是世界上最致命的传染病,是引起死亡的第四大死因,2019年致全球近260万例死亡。2019年上呼吸道感染的发病率高达52.0-128.2/10万。相对于全球而言,我国的感染性疾病诊疗水平偏低,与发达国家差距较大。
传统微生物检测通过分离培养、鉴定、药敏实验等步骤,形成鉴定以及治疗建议,技术成熟处于发展稳定期,但由于步骤多、操作复杂,大大限制了在临床的使用。PCR、质谱和NGS等新技术具有高通量、高精度等优势,对传统检测技术形成极大的补充,临床应用需求广泛。 2021年批准注册1项伴随诊断检测试剂 系罗氏的抗PD-L1 (SP142)兔单克隆抗体试剂(免疫组织化学法)。该产品用于定性检测福尔马林固定、石蜡包埋的组织切片中的程序性死亡配体-1( PD-L1)蛋白表达水平。用于辅助识别采用 TECENTRIQ®(阿替利珠单抗)治疗非小细胞肺癌( NSCLC)的患者,阈值为任何染色强度的≥ 50% TC( PD-L1表达肿瘤细胞百分比)或任何染色强度的≥ 10% IC( PD-L1 表达肿瘤浸润性免疫细胞占肿瘤区域面积的比例)。
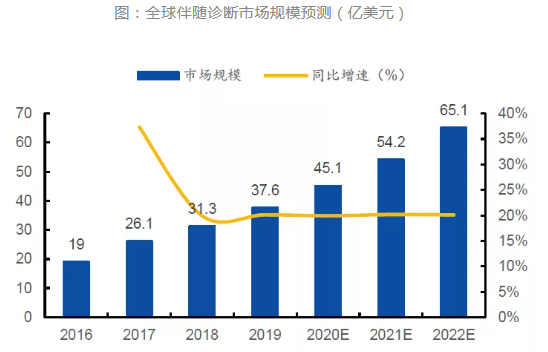
伴随诊断(companion diagnostics,CDx)是一种指导用药方案的体外诊断技术(in vitro diagnostics,IVD),通常与靶标药物一同研发,通过对特定的生物标记物((biomarker)进行检测,筛选最有可能针对该靶标药物产生响应的患者群体,从而改善治疗预后(疗效、风险等)并减少患者的医疗开支。与化学疗法相比,靶向疗法和免疫疗法可以达到更好的治疗效果,同时避免了潜在的严重副作用,正日益成为癌症治疗市场中的重要手段。伴随诊断产品主要基于免疫组织化学法、PCR、FISH、NGS等方法开发,截至2021年底,FDA已批准49个伴随诊断产品,其中绝大多数是用来为肿瘤的用药进行伴随检测。国内首个伴随诊断试剂是2018年1月艾德生物获批的EGFR基因突变检测试剂盒。 据Markets and Markets测算,2017年,全球伴随诊断市场规模为26.1亿美元,预计2022年将达到65.1亿美元,2016年到2022年的年复合增长率将达到22.78%。随着靶向药物的不断开发和人们对健康需求的日益增长,伴随诊断行业正处于高速发展阶段。
参考来源: 1. 广证恒生 新技术突破传统微生物检测瓶颈,NGS引领变革 2. 八阙投研 伴随诊断行业概况 3. bioSeedin柏思荟 靶向药物的黄金搭档:伴随诊断综述 |

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号