CRISPR免扩增系列之(3):E-DNA,基于电化学原理增敏CRISPR诊断检测信号(顺式切割)
2021-9-6 10:26|
发布者: 沙糖桔|
查看: 4056|
评论: 0|来源: CRISPR分子诊断 | 作者阿Q说
摘要: 昨天我们刚刚学习了来自凯斯西储大学的Yifan Dai和Chung Chiun Liu教授研究团队开发的E-CRISPR电化学核酸检测技术,可以提高CRISPR诊断的检测灵敏度。今天我们再来解读一篇同一团队后面开发的新一代电化学检测系统— ...
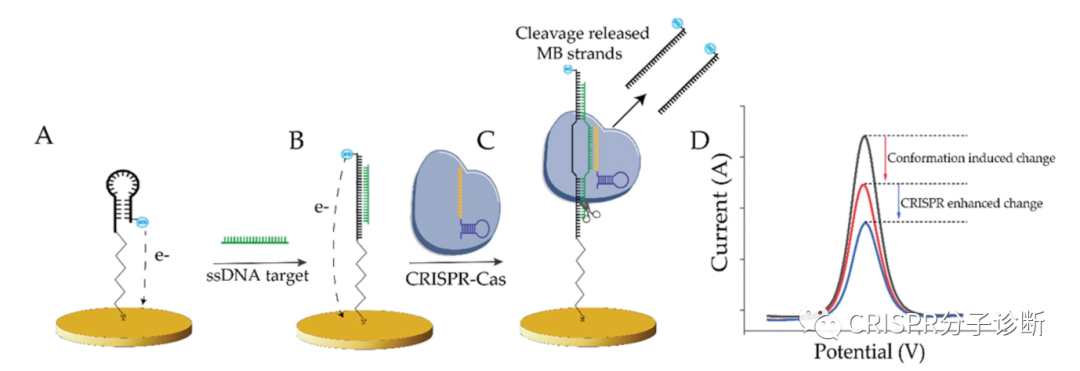
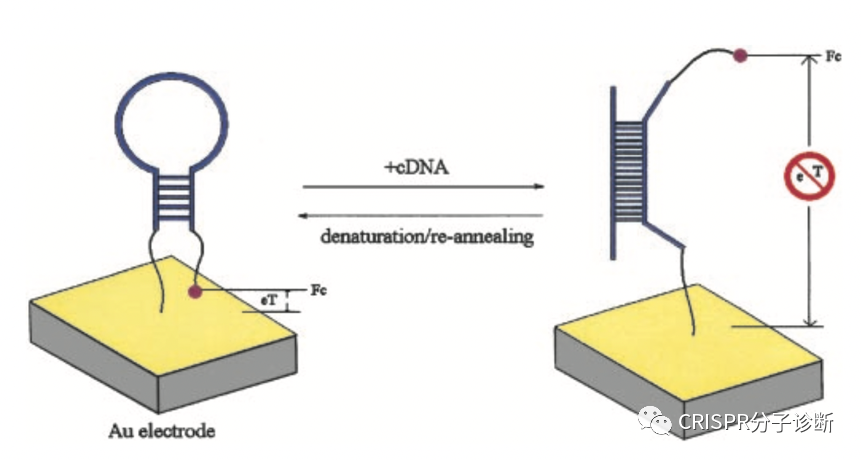
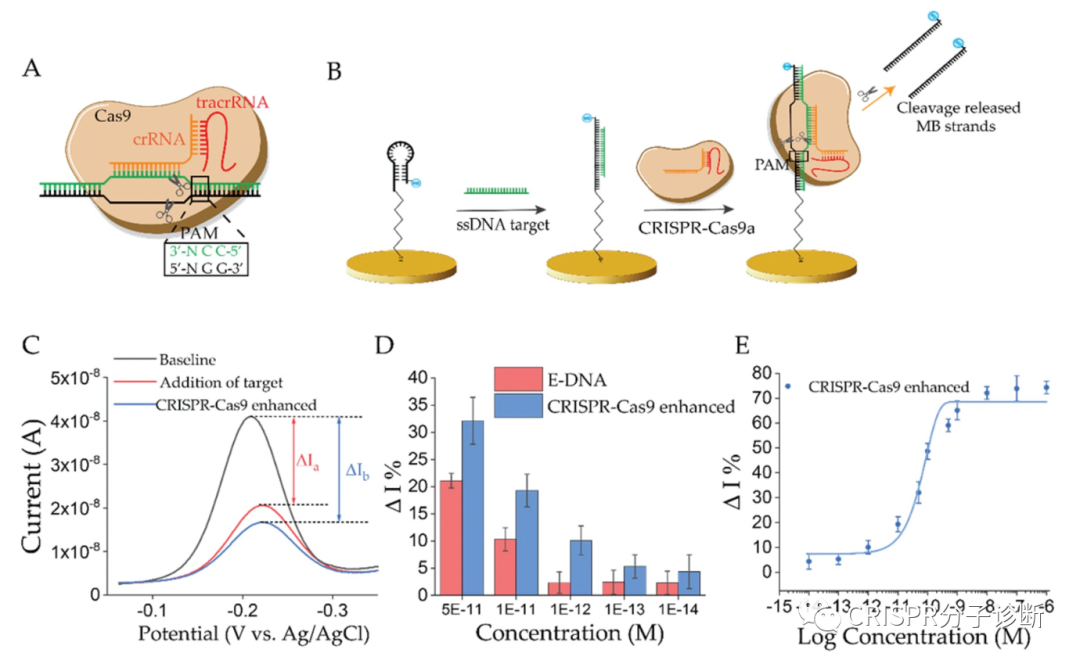
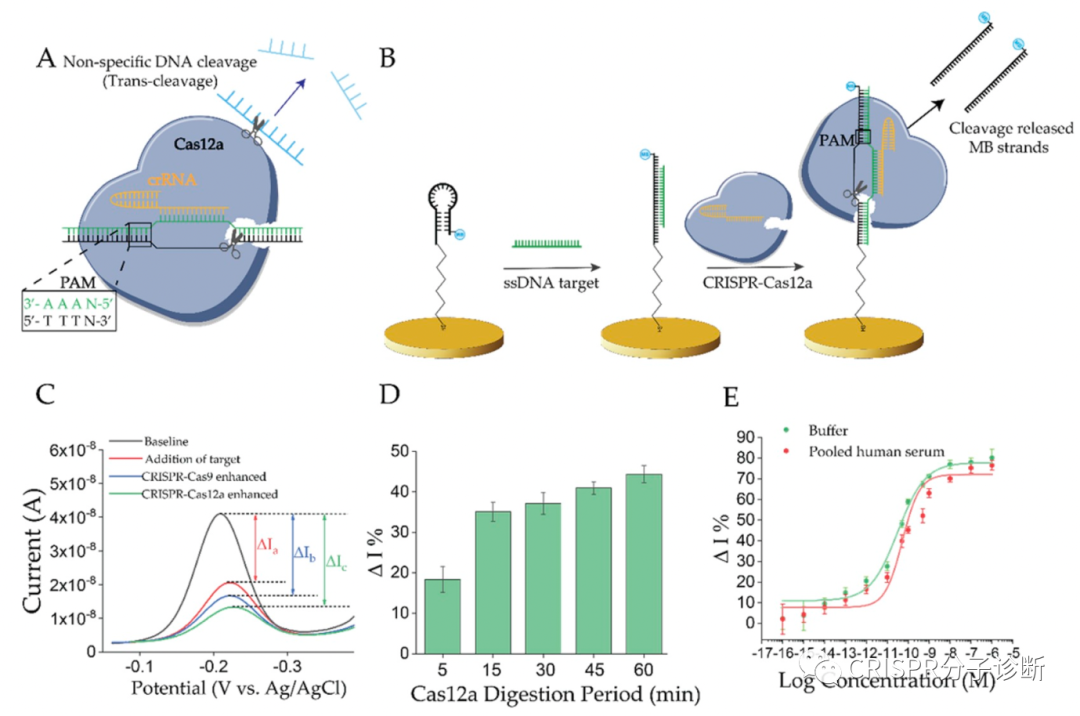
| 昨天我们刚刚学习了来自凯斯西储大学的Yifan Dai和Chung Chiun Liu教授研究团队开发的E-CRISPR电化学核酸检测技术,可以提高CRISPR诊断的检测灵敏度。今天我们再来解读一篇同一团队后面开发的新一代电化学检测系统——E-DNA,又提供了一个新的思路来基于CRISPR的电化学检测靶标核酸。如上图所示,这里发明的E-DNA在原理上和之前2003年Chunhai Fan等人开发的E-DNA系统不太一样,这里E-DNA系统也被称为CRISPR增强的E-DNA技术。之前的E-DNA技术(如下图)主要是基于一种茎环结构将二茂铁(ferrocene)连接在Au电极上,在靶标cDNA存在的情况下,与茎环中的环序列杂交,撑开茎结构,使得二茂铁远离Au电极,引起电子转移效率的变化;该方法检测靶标核酸的灵敏度可以低至10 pM(详见PNAS, 2003, 100(16): 9134-7)。相比之下,CRISPR增强的E-DNA系统(上图所示,以下简称cE-DNA)则是利用CRISPR技术将电子受体切除来造成电子转移效率的差异。首先通过一条与靶标核酸互补的ssDNA序列将电子受体亚甲基蓝连在金电极上;当靶标ssDNA存在时,其便会与连接电子受体的互补ssDNA形成dsDNA,进而被CRISPR-Cas/sgRNA识别并切断,从而使得电子受体脱离金电极,造成电流的变化(注:cE-DNA中连接电子受体的ssDNA会被设计成茎环结构,与靶标互补的序列位于环结构处,其与靶标序列结合后也会引进电流的变化,可供检测;其实也就相当于最初的E-DNA;这个特性在后续的实验中也用到了。) (于2003年发表在PNAS的E-DNA系统示意图)为了验证cE-DNA体系,作者首先利用Cas9进行了测试,证明cE-DNA可以特异地检测靶标ssDNA核酸(Parvovirus B19),且在免扩增的情况下,cE-DNA的检测灵敏度可以达到pM级别(IC50=66.1 pM,这里的IC50应该是和上一篇我们解读的意思一样,可以反映检测的灵敏度,其值越小,灵敏度越高)。 紧接着,作者又证明cE-DNA也可以利用Cas12a来检测靶标核酸,其原理一致,只是利用Cas12a来切割形成的dsDNA序列,释放电子受体,进而引起电流变化以便进行检测。经过测试,Cas12a的IC50可以低至31.9 pM,并可以直接检测人血清中的靶标核酸,其IC50也可以达到51.6 pM。因此,基于Cas12a的cE-DNA系统可以直接用于临床样本的检测(Cas9的是否可以检测血清样本,文章中并没有交待),而且其检测灵敏度要高于Cas9(即31.9 vs 66.1 pM)。 此外,基于Cas12a的cE-DNA技术还可以区分单碱基差异。如下图A,作者首先证明用经典的E-DNA(即上面提到的2003年的那篇PNAS的技术)无法区分单碱基差异;而图B则显示了cE-DNA可以很好地区分PAM区及离PAM区较近区域的位点突变(MT1和MT2),这说明cE-DNA方法有更好的检测特异性(注:并不是说每条靶标序列的突变位点都能被很巧地设计在PAM区,只有突变后形成TT或者破坏TT的突变才可以;另外,对于Cas9来说,有多篇文献证明靠近PAM的位点更重要,但是Cas12a的规律应该是还不太清楚;从目前已经发表的文献来看,应该还是没有总结出什么经典的规律;所以,对于点突变的检测来说,可能还是需要多设计一些crRNA,多测试,相信以后做多了,一定能够总结出规律来)。其次,作者还通过计算靶标DNA的结合和CRISPR的切割之间的相关常数(=结合信号/切割信号;注:上文提到的靶标结合和切割都分别会引起信号变化),来证明cE-DNA能够区分靶标核酸的点突变,主要是归于Cas12a的选择性切割的特性。如果野生型序列的常数为0.9,说明结合后形成的dsDNA基本都被Cas蛋白切割了,且还多切了部分未形成dsDNA的linker ssDNA(正常来说,应该是大约1,这里应该是由于Cas12a的trans-cleavage活性所致);而突变序列的常数则为11.7,说明不到10%的dsDNA(有突变后导至形成bubble的dsDNA)被Cas12a切断,其余野生型序列的0.9存在显著性差异,再次证明cE-DNA方法可以用于靶标核酸的检测(猜想:10%的比例应该还可以进一步降低,比如说通过优化crRNA探针设计,将比例降至5%,1%或更低)。点评:和我们昨天介绍的E-CRISPR免扩增电化学体系不同,上一篇还是利用了Cas12a的反式切割活性来切断金电极上连接的ssDNA,使得电子受体解离,进而引起电流的变化;而这一篇虽然也是利用电化学和CRISPR技术,但是主要利用了Cas9和Cas12a的顺式切割活性,这是我们介绍CRISPR-Dx以来的第一篇非反式切割的文献,倒不是因为这篇工作有多经典,主要是因为这个工作和昨天的工作都是由同一个课题组完成的,为方便起见就放到一起介绍了,明天还是继续介绍反式切割的工作。不过,虽然CRISPR-Dx领域的很大一部分工作都是基于Cas12和Cas13的反式切割活性,倒是也有一部分是基于Cas蛋白的顺式切割活性或dCas蛋白的靶标特异性结合的特性,其中也不乏一些经典,后续会找时间来专门介绍(要专门介绍的貌似有点多啊,大家也可以给我留言,我也可以根据大家的要求来排一下解读的顺序)。这篇工作有几个小创新点,包括整合了E-DNA技术,电化学传感技术,Cas蛋白顺式切割技术等,整体上来说,这篇工作从技术原理上的创新性不是特别高。包括这里证明了cE-DNA系统可以区分靶标核酸的点突变,不过鉴于Cas蛋白的特异性靶标识别的优点,这个结果也属预料之中。此外,cE-DNA还需要使用蛋白酶K对反应后的体系进行酶解反应,切除体系中各种蛋白以防止其非特异性地吸附于电极上而干扰电流信号,这也在一定程度上导至了操作上的繁琐。对于cE-DNA的改进,作者在论文最后也提到了两点:(1)能不能将现在的signal-off改为signal-on,就是说在靶标核酸存在的情况下,才会产生信号,这样有望进一步提升检测灵敏度。(2)根据之前Jennifer的工作(Nature, 2014, 516 (7530): 263–266)显示Cas9结合靶标核酸的亲和力就在pM级别(注:简单看了一下,Cas9/sgRNA与不同靶标的结合力不同,其中最好的一个靶标的结合力为36 pM;大家感兴趣的话可以仔细读下这篇Nature的工作),这因此也限制了cE-DNA的检测灵敏度。所以,作者便提出以后能不能进一步提升Cas蛋白与靶标核酸的结合力?这的确是制约cE-DNA或类似方法检测灵敏度的关键因素之一。完全同意作者提出的这两点。对于如何提高Cas蛋白与靶标核酸的结合力,这可能也是提升Cas反式切割检测灵敏度的关键。不过,对于Cas12a的结合力,不知道是否有人也做过(没仔细研究过,大家看到相关工作的话,欢迎留言提醒);如果没有做过的话,倒是非常值得研究一下。另外,如何才能提高Cas12a对靶标的结合力ne ?欢迎大家留言来一起探讨。 |
声明:
1、凡本网注明“来源:小桔灯网”的所有作品,均为本网合法拥有版权或有权使用的作品,转载需联系授权。
2、凡本网注明“来源:XXX(非小桔灯网)”的作品,均转载自其它媒体,转载目的在于传递更多信息,并不代表本网赞同其观点和对其真实性负责。其版权归原作者所有,如有侵权请联系删除。
3、所有再转载者需自行获得原作者授权并注明来源。

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号