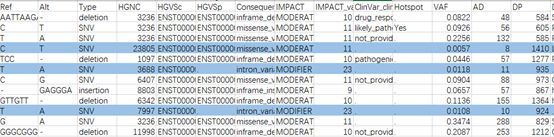
二代测序(Next Generation Sequencing, NGS)是一个强大的功能平台,它可以同时给数以万计的DNA分子进行测序。由于这种可以多个样本同时测序的能力,在个性化医疗、遗传疾病和临床诊断等方面,二代测序也就是高通量测序开创了革命性的领域。但是,对于癌症分子诊断、治疗和监测等需要检测低频基因变异的领域,二代测序错误是一种可能影响变异检测结果的关键混杂因素。 我们常用的体细胞突变(somatic mutation)检测软件包括:mutect2、strelka、varscan等。那么对于突变检测,我们还有哪些要注意呢? 一个典型的NGS工作流程包含多个步骤,包括样品处理、DNA提取、PCR扩增、上机测序。这些步骤中的每一步都可能引入错误。例如,样品处理过程中的DNA损伤可引起C>A/G>T错误,DNA提取或片段化过程中甲基化胞嘧啶自发脱氨成尿嘧啶可引起C>T/G>T错误,另外,目标富集PCR及测序步骤也会引入一些错误。 在体细胞突变检测中,你是否会注意一些异常现象,偶尔会发现一些突变被质控掉了,但这是个例吗?回顾数据会发现,咦?好像同批次个别样本也会有这个突变?例如序列CAGCCGCATCCACCGGTAGCTCTTCTTCTTCTTGCGCT,红色是deletion区域。当然,这个突变被质控掉了,变异检测软件标记的str_contraction。但你是否有看数据的习惯呢?仔细观察突变附近的基因组序列发现该缺失有四个重复单元,是短串联重复区域(STR区域)的特征,这与注释也相符。我们知道,STR检测一般会有滑脱现象,等位基因数值越大(重复的单元数越多)滑脱越多,而等位基因数小的理论上滑脱的可能性很低。 表1 异常体细胞突变示例 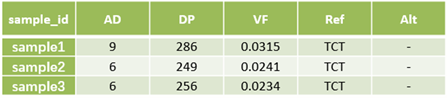
并且对于不同的基因组版本都存在这个现象。另外,由于该突变在同一批数据的多个样本中发现,突变频率较低。到底是捕获探针(原材料)的问题呢?还是STR区域的问题?还是NGS手段的问题?重新设计多重PCR引物,对样本进行检测发现与捕获探针一致,对NGS各个平台、公司的数据回溯分析,包括肿瘤样本和正常样本,发现超过20%的数据(正常对照)包含这个位点,而且频率都比较低(1%上下浮动)。 表2 不同公司不同产品不同测序平台分析 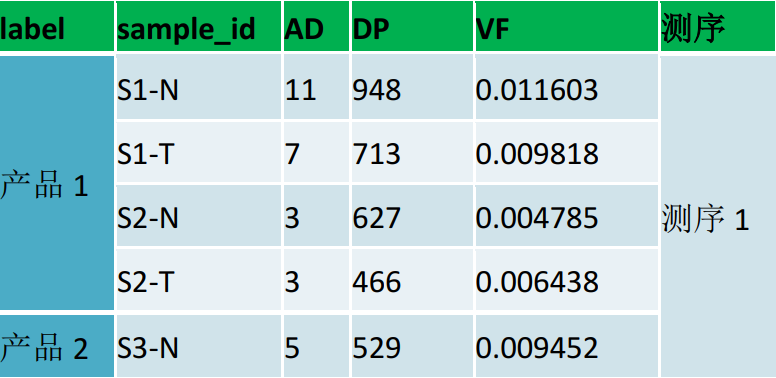
因此判定这个位点是NGS系统性的误差。那就要考虑基因STR区域本身的特点造成的,STR到底是个什么东东呢,为什么会有这种诡异的影响?让我们来解开它神秘的面纱。短串联重复序列(Short tandem repeats, STR),又称微卫星DNA(Microsatellite DNA),通常是基因组中由1~6个碱基单元组成的一段DNA重复序列,是广泛存在于真核生物基因组中的核苷酸重复序列。由于重复单位及重复次数不同,构成了STR基因座的遗传多态性。每个STR由结构包括:具有短重复单元的核心区、核心区两侧保守的侧翼区。 
图1 STR结构示意图
![]()
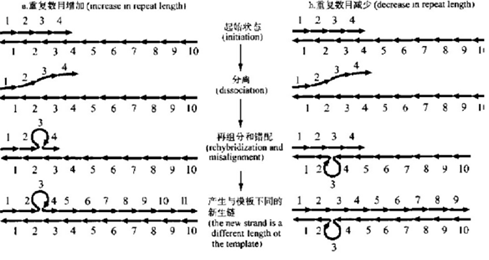
图2 微卫星的滑链错配突变模型 目前主要认为滑链错配是短串联重复序列突变的主要机制。
在DNA复制合成的过程中,新生链和模板链之间在微卫星重复区域可能发生错配,使得一个或者几个重复单位形成环状,未能参与配对。如果未配对的重复单位位于新生链,则最终得到的新生链未配对重复单位数目比模板链多。反之,如果未配对的重复单位位于模板链,则最终得到的新生链未配对重复单位数目比模板链少。有丝分裂过程中DNA链之间的错配引起的复制滑动被认为是导至STR产生的较为常见原因,并且依据不同复制单元大小的不同以及不同物种之间,复制滑动发生的概率也不相同。滑移率与STR的表观突变率不同。体外实验表明,DNA滑移率非常高。但在体内,大部分DNA环被错配重配对系统识别和消除。结果表明,功能性错配修复系统可使STR突变率降低100~1000倍。因此,观察到的STR突变率取决于滑移率和修复系统纠正不匹配的效率。常用的STR基因座突变率在0.1%~0.5%。我们再来分析上面案例,是GATK等变异检测软件中对变异的一种过滤注释,在GATK中称为hard filter,clustered_events的意思是多个变异近距离聚集出现的现象,一般来说clustered_events是一种不真实变异信号。比如可能是同源序列引起的假突变。图3 CNV引起的clustered_events
体细胞检测我们通常会采用配对样本分析,但对于这种假突变,正常对照并不一定能完全测到这些位点,因此很多背景是无法去除点的,这时,构建一组正常样本库(Panel of Normals,PON)对突变进行过滤,就显得很重要了。如果某个突变在这一组正常样本中以一定的较高频率出现,就认为这个突变为系统错误引起的,可以进行过滤,例如下图中所有位点均为case-control方式检测出的位点,底色为白色的行表示真实位点,其余位点均为非真实位点,没有被case-control方式过滤掉,蓝色表示可以被PON库过滤掉的位点。
图4 PON库比control多过滤掉的假突变示例
再回到clustered_events这块,如上面图3中的突变真实并不存在,数据库中没有记录,有可能是基因组上相似区域的序列同时比对到同一位置产生,被标记为clustered_events,可以按照指标被过滤掉;而下面图5中的突变是真实存在的,在COSMIC数据库中有记录,虽然标记为clustered_events,但是被默认指标过滤掉。
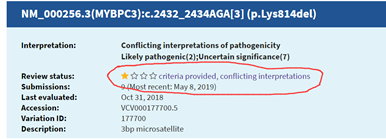
图5 MNP识别成clustered_events回到这个位点,我们再看下clinvar、dbsnp、cosmic都有记录。对于胚系突变,clinvar中有记录但结论冲突,ExAC等数据库也有但非常罕见,dbsnp中每个记录都没有验证信息,也并没有allele balance信息。对于体细胞突变,cosmic中有但信息也不多。这个位点是否值得怀疑?当然,并不排除真的有样本是存在这个突变,大家可以探讨。如果按照默认的软件指标对变异进行过滤可能会造成假阳或假阴,也就是造成检测错误,而且错误还可能来源于基因组组装错误(干试验+湿实验)。所以软件质控并不是万用的,对于体细胞突变检测,即使是配对样本,我们也需要PON,而对于假阴性,我们同样需要累积(包括cosmic公共数据库使用和内部积累),平台的数据累积才是最合适的质控。临检的分析虽没有动植物科研的多样化手段和各种美图显得复杂,但是我们对准确度的要求非常高,开展检测一定要慎重。
Ma X, Shao Y, Tian L, et al. Analysis of error profiles in deep next-generation sequencing data[J]. Genome biology, 2019, 20(1): 50.Temple-Smolkin R L, Voelkerding K V, Nikiforova M N. Guidelines for Validation of Next-Generation SequencingeBased Oncology Panels[J]. 2017.
| 
 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号