金桔
金币
威望
贡献
回帖0
精华
在线时间 小时
|
登陆有奖并可浏览互动!
您需要 登录 才可以下载或查看,没有账号?立即注册


×
简介
在如何自学生物信息学?这个问题的最高赞回答下,树嘉哥给我们推荐了《bioinformatics data skills》这本入门书籍。我自己3年前入门生信时,也是看的这本书,受益良多。在这里推荐给大家。推荐的理由有3点:1. 这本书对新手非常友好,学到的东西大多马上可以投入到实战当中。2. 教会你一些良好的习惯,大量节省以后花在踩坑上的时间。3. 作者公布了代码和数据,可以直接在github上下载练习(链接:https://github.com/vsbuffalo/bds-files)。
主要内容
- 良好的分析习惯(第一章)
- 操作系统(linux)、编程语言(shell,R)和常用工具(awk, grep, sed)等(第二章到第八章、第十二章到第十三章)
- 常见数据类型(gff, fasta, sam等)的处理(第九章到第十一章)
在我看来,第一章是这本书的特色。很多关于生信入门的帖子里面都没有提到。主要是强调数据分析的稳健性(robust)可重复性(reproducible)。其他工具类的知识都可以用到再学,但是这两个概念很有必要在一开始的时候就吃透。其带来的益处,就好比你小时候就理解了“温故而知新”的道理,并不断地实践带来好处一样。
因此,在这里,我把原著的导读部分和第一章整理了出来,如果大家感兴趣,可以去把原著找来读读,相信你会喜欢上的。
<hr/>
序
这本书是我两年前问自己的一个问题的答案:“当我开始学习生物信息学时,我想先读哪本书?”“当我开始在这个领域工作时,我在python和r方面有编程经验,但其他方面没有。我到处寻找一本关于生物信息学的很棒的入门书,虽然我找到了一些好的书,但大多数书并没有针对我作为一名生物信息学家所做的日常工作。我发现的一些文献从理论和算法的角度探讨了生物信息学,涵盖了诸如Smith-Waterman比对、系统发育重建、motif发掘等主题。虽然它们读起来很吸引人(我确实建议你研究一下这篇材料),但在我的日常生物信息学工作中,我不需要从头开始实现生物信息学算法,这些算法的许多出色、高度优化、测试良好的实现已经存在。其他的生物信息学文本采取了一种更实用的方法,引导不熟悉计算的读者完成每一步的任务,比如运行一个比对工具或从数据库下载序列。虽然这些更适用于我的工作,但这些书的大部分材料都过时了。
正如你可能猜到的,我找不到最好的“第一本”生物信息学书籍。《生物信息学数据技能》是我正在寻找的书的版本。这本书的目标读者不确定如何在了解脚本语言和实践生物信息学之间弥合巨大的鸿沟,以一种稳健和可重复的方式回答科学问题。为了弥合这一鸿沟,人们必须学习数据技能——一种使用核心工具集来操作和探索在生物信息学项目中遇到的任何数据的方法。
数据技能是学习生物信息学的最佳方式,因为这些技能使用经过时间考验的开放源代码工具,这些工具仍然是操作和探索不断变化的数据的最佳方式。这种学习方法经受住了时间的考验:高通量测序的出现迅速改变了生物信息学这个领域,但这些相同的工具和技能对新的数据仍然适用。毕竟,下一代数据只是数据(不同的数据,以及更多的数据),而生物信息学家掌握了将他们的工具应用于新数据解决问题的基本技能。生物信息学数据技能是为您提供这些核心工具的培训,并帮助您发展这些相同的技能。
这本书的理念
许多以生物信息学为起点的生物学家倾向于将“学习生物信息学”等同于“学习如何运行生物信息学软件”。这是一个不幸的事实。这类似于认为“学习分子生物学”只是“学习移液”。除了在第11章中用于生成数据的几个简单例子外,本书没有涉及运行生物信息格式学软件,如比对工具、组装工具或变异检测工具。运行生物信息学软件并不那么困难,不需要太多技能,也没有体现任何生物信息学的重大挑战。我不教如何在生物信息学数据技能中运行这些类型的生物信息学应用程序,原因如下:
- 你很容易可以自己搞定。
- 当新版本发布或者新的软件推出时,这些学习材料就会过时。
- 作者写的文档往往是最好的学习材料。
相反,这本书的方法是集中在技能生物信息学家用来探索和提取意义复杂,大型生物信息学数据集。从这些数据集中探索和提取信息是生物信息学研究的有趣部分。生物信息学数据技能的目标是教你需要的计算工具和数据技能,你可以随意探索这些大型数据集。这些数据技能给了你自由;你将能够查看任何数据库中的任何生物信息学数据,以及任何大小的文件,并开始探索数据以提取生物意义。
贯穿本书,我强调以一种稳健和可重复的方式工作。我相信,在现代计算工作中,可重复性和稳健性这两个特性经常被忽视。我所说的稳健,是指您的工作能够抵御无声错误(slient error, 程序可以正常输出结果,但输出的结果是错的)、干扰因素、软件错误和混杂的数据。相反,脆弱的方法并不能降低某些类型错误对结果产生不利影响的几率。可重复的意思是你的工作可以被其他研究人员重复,他们可以得到相同的结果。要做到这一点,您的工作必须有良好的文档记录,并且您的方法、代码和数据都需要可用,以便其他研究人员拥有复制每个步骤的材料。可重复性还取决于您的工作是否稳健,如果在不同的机器上运行的工作流产生不同的结果,那么它既不稳健也不完全可再现。在第2章中,我更深入地介绍了这些概念,这些主题在整本书中再次出现。
为什么这本书关注测序数据?
生物信息学是一门广泛的学科,涉及蛋白质组学、代谢组学、结构生物信息学、比较基因组学、机器学习和图像处理等子领域。《生物信息学数据技能》主要集中在处理序列数据上,原因有几个。
首先,测序数据丰富。目前,没有其他的“omics”数据像高通量测序数据那样丰富。测序数据在生物学中有广泛的应用:变异检测和基因分型、基因表达研究的转录组测序、蛋白质-DNA相互作用分析(如Chip Seq)和甲基化研究的亚硫酸氢盐测序(仅举几个例子)。用测序数据来回答生物学问题的方法只会继续增加。
第二,测序数据对于提高您的数据技能是非常好的。即使您的目标是在将来分析其他类型的数据,测序数据也可以作为学习的很好的示例数据。开发处理连续数据所需的文本处理技能将适用于处理许多其他数据类型。
第三,生物信息学的其他子领域更具领域特异性。测序的广泛可用性和不断下降的成本使得来自所有学科的科学家能够利用基因组学数据来回答他们系统中的问题。相比之下,蛋白质组学或高通量图像处理等生物素形成子学科更为专业化,也不那么广泛。不过,如果你对这些领域感兴趣,生物信息学的数据技能将教会你有用的计算和数据技能,这将有助于你的研究。
这本书的受众
根据我在加州大学戴维斯分校(UC Davis)为期一周的强化课程中向朋友、同事和学生教授生物信息学的经验,大多数希望学习生物信息学的人要么是生物学家,要么是计算机科学家/程序员。生物学家希望发展分析自身数据所必需的计算技能,而程序员和计算机科学家则希望将其计算技能应用于生物逻辑问题。尽管这两个群体在生物学知识和计算经验上存在很大差异,但生物信息学数据技能涵盖了对两者都有帮助的材料。
如果你是一名生物学家,《生物信息学数据技能》将教会你使用生物信息学数据所需的核心数据技能。重要的是要注意,《生物信息学数据技能》并不是一本如何进行生物信息学的书;这样一本关于生物信息学的书很快就会过时,或者过于专注于帮助大多数生物学家。读完这本书,你还需要补充与你的具体研究和系统,以及现代统计和生物信息学方法,你的子领域使用的知识。例如,如果你的项目涉及到比对reads到参考基因组,这本书不会告诉你最新和最好的适用于你的操作系统的比对软件。但是,无论您使用哪种比对软件,您都需要对比对格式以及如何操作比对数据有一个透彻的了解——这是第11章涉及的主题。在这本书中,这些通用的计算和数据技能应该是一个坚实的、广泛适用的基础,而且大多数生物学家可以建立这些基础。
这本书的难度
《生物信息学数据技能》被设计成一本全面的、干货满满的书。当我开始写这本书的时候,我确信我能做的最大的错误就是把生物信息学过度简化。作为一名专业的生物信息学家,我经常看到一些非常微妙的问题会突然出现,并在没有被发现的情况下对分析结果产生不利影响。我不希望你在看完这本书后仍犯这样错误。我在生物信息学数据技能方面所涉及的主题的深度是为了让你在自己的工作中能够抓住类似的问题,从而使你的结果是可靠的。
结果是,这本书的某些章节对一些读者来说相对较难。别灰心!像大多数科学一样,这种材料很难理解,可能需要读几遍才能完全理解。在整本书中,我试图指出哪些章节比较难,以便您可以跳过这些部分,稍后再返回。
最后,我经常在整本书中使用技术术语。我不喜欢用行话,但有必要在计算中交流技术概念。它主要帮助您搜索其他资源和帮助。“left outer join”比“一个表中包含空记录的数据合并”更容易被谷歌成功搜索。
这本书假设你已经满足以下条件
《生物信息学数据技能》是帮你过渡到生物信息学的桥梁。为了确保每个人从同一个起点出发,这本书从几个简单的章节开始。在第2章中,我介绍了建立生物信息学项目的基础知识,在第3章中,我教授了一些Unix相关的知识,旨在确保您对Unix有一个扎实的理解(因为Unix在后面的章节中是一个很大的组成部分)。不过,作为一本过渡性的书,我对你做了一些假设:
你懂一门脚本语言。这是这本书最大的假设。除了一些Python程序和R材料(R在第8章中介绍),本书并不直接依赖于使用大量脚本。然而,在学习脚本语言时,您已经遇到了许多重要的计算概念,例如使用文本编辑器、在命令行上运行和执行程序以及基本编程。如果您不了解脚本语言,我建议您在阅读本书时学习Python。诸如Mitchell L.Model(O'Reilly,2009)、Learning python(Mark Lutz,2013)第5版和Alex Martelli(O'Reilly,2006)的《python in a nutshell,2nd》等书都是很好的入门之作。如果您知道的脚本语言不是Python(例如Perl或Ruby),那么你应该可以看懂大多数示例(尽管您需要将一些示例翻译成您选择的脚本语言)。
你会用一种文本编辑器。了解文本编辑器(例如Emacs、Vim、Text-Mate2或Sublime文本)的方法是很重要的。使用文字处理器(如Microsoft Word)将不起作用,我不鼓励使用像记事本或OS X的Tex-Tedit这样的编辑器,因为它们缺乏语法高亮对常用编程语言的支持。
你懂基本的Unix命令行操作。例如,我假设您知道terminal和shell之间的区别,了解如何输入命令,什么是options/flags和arguments,按“Up”键来快速输入之前输入过的指令。您还应该对Unix文件层次结构有基本的了解(包括home目录、相对目录和绝对目录以及根目录等概念)。您还应该能够在Unix中使用cd、ls、pwd、mv、rm、rmdir和mkdir等命令移动和操作目录和文件。最后,您应该基本了解Unix文件的所有权和权限,并使用chown和chmod更改这些权限。如果这些概念不清楚,我建议您先在Unix自己操作一下(小心!),这一步骤可以参考一本很好的入门书籍,如Steven Haddock和Casey Dunn(Sinauer,2010年)的《Practical Computing for Biologists》,或Keith Bradnam和Ian Korf的《UNIX and Perl to the Rescue》(剑桥大学出版社,2012年)。
你有基本的生物学知识。《生物信息学数据技能》需要你自带生物学知识。这里面的例子不需要太多的生物学背景,除了DNA、RNA、蛋白质和基因,以及分子生物学的中心法则。您还应该熟悉一些非常基本的遗传学和基因组学概念(例如,单核苷酸多态性、基因型、GC含量等)。书中所有的生物例子都设计得非常简单;如果你对任何话题都不熟悉,你应该能够快速浏览维基百科的文章并继续这个例子。
你对正则表达式有基本的了解。偶尔,我会用到这本书中的正则表达式。在大多数情况下,我尝试快速地逐步了解正则表达式的工作原理,这样您就可以得到一般的概念。如果在学习脚本语言时遇到了正则表达式,那么就可以开始了。如果没有,我建议你学习基础知识——不是因为正则表达式在书中被大量使用,而是因为掌握正则表达式是生物信息学中的一项重要技能。Michael Fitzgerald(O'Reilly)的《Introducing Regular Expressions》是一个很好的向导。如今,借助http://regex101.com和http://www.debuggex.com等在线工具,编写、测试和调试正则表达式比以往任何时候都容易。我建议在您自己的工作中使用这些工具,并在逐步执行正则表达式示例时使用这些工具。
你懂得如何获得帮助和阅读文档。在这本书中,我尽量减少可以在手册、帮助文档或在线上找到的教学信息。这有三个原因:
- 我想节省空间,专注于以其他地方找不到的方式展示材料。
- 手册页和文档将始终是获取这些信息的最佳资源
- 在文档中快速找到答案的能力是学习计算机技术时所能培养的最重要的技能之一。
最后一点特别重要;您不需要记住命令或r函数的所有参数,只需要知道在哪里可以找到这些信息。编程人员在工作中经常查阅文档,这就是为什么存在诸如man(在UNIX中)和help()(在r中)这样的文档工具。
你能管理你的计算机系统(或者有一个计算机管理员)。这本书没有教你系统管理技能,如建立一个生物信息学服务器或集群,管理用户帐户,网络安全,管理磁盘和磁盘空间,RAID配置,数据备份和高性能计算概念。根本没有足够的空间来涵盖这些重要的主题。然而,如果你没有一个系统管理员,并且需要亲自为你的实验室或研究小组担任这个角色,那么这些都是非常非常重要的,你也必须掌握这些技能。坦率地说,系统管理技能需要数年才能掌握,而优秀的系统管理员在处理问题方面拥有令人难以置信的耐心和经验,这些问题会使大多数科学家变得疯狂。如果有条件的话,你可以雇佣一个在实验室或组之间共享的全职系统管理员,或者使用一个带有系统管理员的大学集群。最后,这不需要说,但以防万一:不断备份数据和工作。在学习Unix的过程中,一个命令就能让你丢失重要的文件,所以定期备份是防止这种情况的最佳保护方式。
GitHub上的补充材料
本书示例所需的补充材料可在Git-Hub存储库中找到。您可以根据需要从此存储库(https://github.com/vsbuffalo/bds-files)下载材料(存储库按章节组织),也可以使用下载zip链接下载所有内容。一旦您在第5章中学习了Git,我建议您克隆存储库,以便在意外覆盖示例文件时恢复它们。
尝试现在导航到此存储库并四处搜索,以便熟悉布局。在序言的目录中,您可以找到README.md文件,其中包括有关我讨论过的许多主题的附加信息。除了本书中所有示例所需的补充文件外,此存储库还包含:
- 记录了如何生成所有补充文件和这些文件从何而来的文档。在某些情况下,我使用了makefile或脚本(这两个主题都在第12章中介绍)来创建示例数据,并且这些资源在每个章节的Github目录中都是可用的。这些材料不仅提供了可重复性,而且还能作为额外的学习材料。
- 扩展阅读材料。读者可能会对某些章节特别感兴趣。这些材料在每个章节的README.md文件中。我还收录了其他资源,比如推荐书单,以便进一步学习。
- 勘误表,以及材料因某种原因过时时的任何必要更新。
我选择在Github上托管生物信息学数据技能的补充文件,这样我就可以更新所有内容并解决读者可能遇到的任何问题。如果您发现本书或其补充材料有任何问题,请随时在Github上创建新问题。
计算资源和设置
这整本书都是在我的笔记本电脑上写的,这是一个15英寸的MacBook Pro,有16GB的内存。虽然这是一台功能强大的笔记本电脑,但它比生物信息学计算中常见的服务器要小得多。所有的例子都被设计和测试来在类似配置的机器上运行。几乎每个示例都应该在内存为8GB的计算机上运行。本书中的所有示例都适用于Mac OS X和Linux——不支持其他操作系统(主要是因为现代生物信息学依赖于基于Unix的操作系统)。本书中所需的所有软件都是免费提供的,并且易于安装;由于需要软件安装,我在每个部分提供了一些基本说明。通常,您应该使用操作系统的包管理系统(例如,在Ubuntu/Debian上使用apt-get)。如果您使用的是Mac,我强烈推荐Homebrew,这是一个非常好的OS X包管理器,它允许您从命令行轻松安装软件。您可以在home-brew的网站上找到详细的说明,最重要的是,home brew维护了一系列称为homebrew-science的科学软件包,包括我们在本书中使用的生物信息学软件。按照homebrew-scienc的README.md中的说明了解如何安装这些科学程序。
这本书的组织方式
这本书由三部分组成:第一部分,包含一个关于分析思维的章节;第二部分,包括开始一个生物信息学项目的基础;第三部分,包括生物信息学数据技能。虽然章节是按顺序编写的,但是如果您对Unix和R比较熟悉,那么您完全可以跳过这些章节。
第一部分
在第一章中,我介绍了为什么通过培养数据技能来学习生物信息学是最好的方法。我还介绍了这本书的思想,并描述了可复制和稳健的生物信息学和一些建议,以适用于您自己的工作。
第二部分
《生物信息学数据技能》第二部分介绍了启动生物信息学项目所需的基本技能。首先,我们将在第2章中介绍如何设置和管理项目目录。这似乎是一个微不足道的话题,但复杂的生物信息学项目要求我们考虑项目管理。在研究的狂热中,到处都会有文件。从一个精心组织的项目开始可以避免将来的很多麻烦。我们还将了解Markdown,这是纯文本项目文档的一种有用格式。
在第三章中,我们探讨了生物信息学要用到的Unix知识。这是为了确保您对基本概念(如管道、重定向、标准输入和输出等)有一个扎实的掌握。了解这些先决条件主题将使您能够专注于分析后面章节中的数据,而不是绞尽脑汁地理解Unix基础知识。
大多数生物信息学任务需要比个人电脑更多的计算能力,这意味着我们必须使用远程服务器和集群。第4章介绍了在使用远程机器时提高生产率的一些技巧和技巧。
在第5章中,我们学习了Git,它是一个版本控制系统,使项目的版本管理变得容易。生物信息学项目充满了大量的代码和数据,这些代码和数据应该使用与协作开发的软件相同的现代工具进行管理。Git是一款功能强大的大型软件,因此这是一个很长的章节。但是,编写本章的目的是为了跳过有关branching的部分,稍后再回头学。
第6章介绍生物信息学项目中的数据:如何下载大量数据,使用数据压缩,验证数据完整性,以及如何为项目重复下载数据。
第三部分
在第三部分中,我们的注意力转向开发所有生物信息学家在日常工作中解决问题所需的基本数据技能。第7章重点介绍Unix数据工具,它允许您快速编写强大的流处理Unix管道来处理生物信息学数据。这种方法是现代生物信息学的基石,是绝对必要的数据技能。
在第8章中,我通过学习探索性数据分析技术来介绍R语言。本章准备使用R来使用可视化(visualization)和数据摘(data summaries)要等技术探索您自己的数据。
基因组范围数据(Genomic range data)在生物信息学中普遍存在,因此我们在第9章中研究了范围数据和范围操作。我们将首先通过不同的方法来表示基因组范围,并使用Bioconductor的IRanges包进行范围操作,以增强我们的范围思维直觉。然后,我们将使用基因组范围处理基因组数据。最后,我们将查看用于在命令行上处理范围数据的BEDTools工具套件。
在第10章中,我们学习了序列数据,这是生物信息学数据的主要部分。我们将研究FASTA和FASTQ格式(及其局限性),并从序列中剔除低质量的碱基,看看这如何影响质量分数的分布。我们还将研究FASTA和FASTQ解析。
第11章重点介绍alignment数据格式SAM和BAM。理解和操作这些格式的文件是处理高通量测序数据的一项不可或缺的生物信息学技能。我们将看到如何使用Samtools来操作这些文件和可视化数据,并通过一个详细的示例来逐步说明变异检测的一些复杂性。最后,我们将学习如何使用Pysam解析SAM/BAM文件,这样您就可以编写自己的脚本来处理这些特殊的数据格式。
大多数日常的生物信息学工作包括编写数据处理脚本和流程。在第12章中,我们将研究如何以一种稳健且可重复的方式编写这样的数据处理流程。我们将专门研究bash脚本,使用诸如find和xargs之类的强大的Unix工具操作文件,最后快速了解如何使用make和make files编写流程。
在生物信息学中,我们的数据往往太大,无法存储在计算机内存中。在第7章中,我们看到了使用Unix管道进行流处理如何有助于解决这个问题,但是第13章介绍了另一种方法:内存不足方法。首先,我们将看到tabix,一种快速访问索引标签分隔文件中信息的方法。然后,我们将通过使用sqlite分析一些GWAS数据来了解SQL的基础知识。
最后,在第14章中,我将讨论下一步你应该去哪里进一步发展你的生物信息学技能。
第一章 如何学习生物信息学
现在,在世界各地的实验室里,机器正在对地球上生命的基因组进行测序。即使成本快速下降,技术进步巨大,基因测序,我们也只能概览到包含在每个细胞、组织、有机体和生态系统中的生物信息。然而,我们收集到的一点点生物信息,就相当于生物学家需要处理的大量数据。在人类历史上,没有其他任何一个时期如此依赖于我们处理和分析数据的技能来理解生命的复杂性。这本书是关于通过发展数据技能来学习生物信息学的。在本章中,我们将看到什么是数据技能,以及为什么学习数据技能是获取生物信息学的最佳方式。我们还将研究稳健和可重复的研究需要什么。
为什么要学习生物信息?——日渐增长的生物学数据
生物信息学家关注的是从大量的数据中获得生物学上的理解,这些数据具有专门的技能和工具。在生物学历史的早期,数据集是小的和可管理的。大多数生物学家在上完统计学课程后,可以在个人台式电脑上使用Microsoft Excel分析自己的数据。然而,这一切都在迅速变化。大型序列数据集分布广泛,而且只会在未来变得更加普遍。分析这些数据需要不同的工具、新的技能和许多具有大量内存、处理能力和磁盘空间的计算机。
在相对较短的时间内,测序成本急剧下降,使研究人员能够利用测序数据来帮助回答重要的生物学问题。早期测序是低吞吐量和昂贵的。全基因组测序工作成本高昂(人类基因组成本约27亿美元),只有通过大规模的合作才能实现。自从人类基因组发布以来,测序成本一直呈指数级下降,直到2008年左右,如图1-1所示。随着新一代测序技术的引进,对一个一百万碱基进行测序的成本下降得更快。在这一关键时刻,一项只有建立大型测序合作(或具有充足资金的单个研究人员)才能负担得起的技术,成为所有生物学研究人员都负担得起的技术。你读这本书很可能是为了学习使用已经大幅降低成本的高通量测序数据。
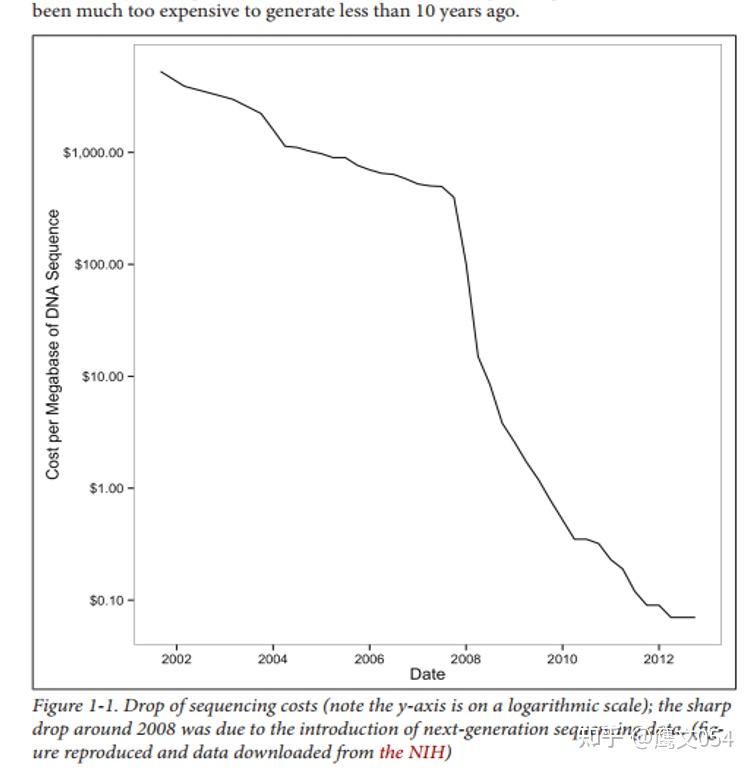
图1-1
由于这些新技术,排序成本下降的后果是什么?正如你所猜测的,大量的数据。在指数增长之后,生物数据库的数据膨胀了。虽然合作者之间共享的小型数据库已经足够了,但是现在全世界的服务器上都有petabytes级别的有用数据。对生物学问题的关键见解不仅存储在硬盘上未分析的实验数据中,而且还在数千英里外数据中心的磁盘上旋转。
生物数据库的增长和测序成本的下降一样令人震惊。以Sequence Read Archive(简称SRA,以前称为Short Read Archive)为例,它是来自测序实验的原始序列数据的存储库。自2010年以来,增长非常显著,见图1-2。
要考量这种难以置信的序列数据增长,可以参考摩尔定律。戈登·摩尔(Intel的联合创始人)观察到,计算机芯片中的晶体管数量大约每两年翻一番。每片芯片上的晶体管越多,计算机处理器的速度就越快,计算机中的随机存取存储器就越多,这就产生了更强大的计算机。这一惊人的技术改进速度每两年翻一番,很可能是人类所见过的最快的技术增长。然而,自2011年以来,存储在SRA中的序列数据量甚至超过了这种难以置信的增长速度,每年都翻了一番。
不断开发的新生物数据分析工具,及其底层算法的改进使情况更加复杂。2012年的综述列出了70多个短读段比对工具(Fonseca等人,2012年;见http://bit.ly/htsmappers)。同样,在过去的五年中,我们的基因组装配方法发生了很大的变化,因为长序列的装配方法(如overlap-layout consensus algorithms算法)随着短高通量序列读取的出现而被放弃。现在,测序化学的进步导致了更长的序列读取长度,新的算法正在取代其他只有几年历史的算法。
不幸的是,生物信息学工具的丰富和快速发展有着严重的缺点。通常,生物信息学工具没有充分的基准,尽管有,也只是对某个有机体的基准。这使得新生物学家很难找到和选择最好的工具来分析他们的数据。为了使事情变得更加困难,一些生物信息学项目没有得到积极的开发,因此它们往往过时了,或者携带了可能对结果产生负面影响的bugs。所有这些都使得在你自己的研究中选择合适的生物信息学程序变得困难。更重要的是,必须严格评估基于您自己的数据运行的生物信息学程序的输出。我们将看到数据技能如何帮助我们评估第二部分中的程序输出的示例。
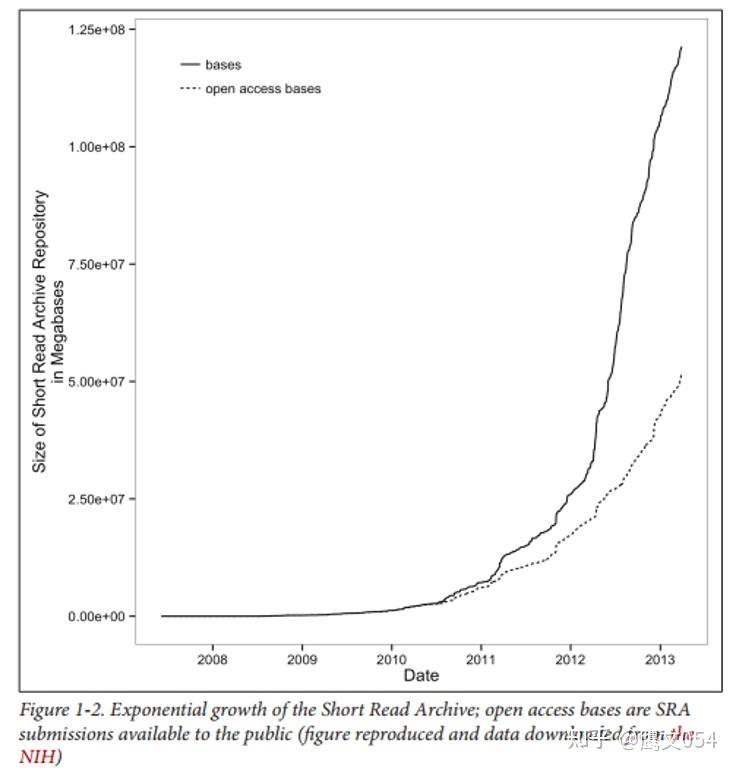
图1-2
通过学习数据技能来学习生物信息
生物数据变化如此之快,你应该如何学习生物信息学?由于所有的工具都在那里,并且不断地被创造出来,生物学家如何才能知道一个程序是否能在其有机体的数据上适当地工作?
解决方法是像生物信息学家那样接触生物信息学:尝试东西,并评估结果。通过这种方式,生物信息学就是拥有使用计算机进行数据实验和理解结果的技能。实验部分很容易,这对大多数科学家来说是很自然的。对大多数生物学家来说,限制因素是拥有在计算机上自由实验和处理大数据的数据技能。这本书的目的是教你必要的生物信息学数据技能,让你在计算机上实验数据,就像你在实验室里做实验一样容易。
不幸的是,许多生物学家常用的计算工具无法扩展到现代生物数据的规模和复杂性。复杂的数据格式、多种程序的接口以及评估软件和数据使得大型生物信息学数据集难以使用。学习核心生物信息学数据技能将为您学习、应用和评估任何生物信息学程序或分析方法奠定基础。在未来的10年里,生物信息学家可能只会用到很少的生物信息学软件。但我们肯定会利用数据技能和实验来评估未来的数据和方法。
那么什么是数据技能呢?它们是一组计算技能,使您能够使用一套常用的工具快速即兴创作一种查看复杂数据集的方法。一个很好的类比就是爵士乐音乐家所说的拥有“肖邦”。一个拥有好肖邦的爵士乐音乐家可以走进一个夜总会,听到一首熟悉的标准歌曲正在演奏,识别和弦的变化,并开始在这些和弦上演奏音乐思想。同样地,一个具有良好数据技能的生物信息学家可以接收到一个巨大的序列数据集,并立即开始使用一套工具来查看数据所讲述的故事。
就像一个掌握了他的乐器的爵士音乐家,一个拥有优秀数据的生物信息学家掌握了一套工具。学习工具是发展数据技能的一个必要步骤,但还不够(同样,学习一种乐器是演奏音乐思想的必要步骤,但还不够)。在整本书中,我们将培养我们的数据技能,从第二部分的生物信息学项目和数据,到第三部分的数据分析小工具和大工具的学习。然而,这本书只能让你走上正确的道路;真正的掌握需要通过反复把技能应用到实际问题上来学习。
可重复与稳健的研究:新的挑战
生物学越来越多地使用大型测序数据集,其变化超过了我们需要的工具和技能:它也改变了我们的科学发现的可重复性和稳健性。当我们利用新的工具和技能来分析基因组学数据时,有必要确保我们的方法与其他任何实验方法一样具有重复性和稳健性。不幸的是,我们的数据规模和分析工作流程的复杂性使得这些目标在基因组学中尤其困难。
再现性的必要条件是我们共享我们的数据和方法。在前基因组学时代,这要容易得多。论文可以包括详细的方法总结和完整的数据集,正如克里特曼1986年的论文中对4713bp adh基因侧翼序列所做的那样(它被嵌入到论文中间)。现在,论文有很长的补充方法、代码和数据。共享数据也不再是琐碎的事情,因为测序项目可以包含Tb级别的相关数据。分析中使用的参考基因组和注释数据集不断更新,这会使再现性变得困难。与期刊网站上补充材料、方法和数据的链接中断,当研究机构移动或更新其网站时,研究机构网站上的材料消失,当开发人员离开而不更新代码时,软件项目变得陈旧。在这本书中,我们将在做实际分析的同时,看看可以做些什么来提高项目的可再现性,因为我相信这些都是必要的补充活动。
此外,生物信息学分析的复杂性可能导致发现容易出错和技术混淆。即使是相当常规的基因组学项目也可以使用几十种不同的程序、复杂的输入参数组合以及许多样本和注释数据集;此外,工作可能会分布在服务器和工作站上。所有这些计算数据处理步骤都会产生用于更高层次分析的结果,从而得出我们的生物学结论。最终的结果是,研究结果可能停留在许多加工步骤的摇晃的支架上。更糟的是,生物信息学工作流程和分析通常只运行一次,以产生出版物的结果,然后再也不会运行或测试。这些分析可能依赖于所使用的所有软件的非常具体的版本,这会使在不同的系统上重现变得困难。在学习生物信息学数据技能时,必须同时学习再现性和健壮的最佳实践。让我们依次看看再现性和健壮性。
可重复的研究
重复科学发现是确认它们准确的唯一方法,而不是单个实验或分析的人工产物。卡尔·波普尔在《科学发现的逻辑》一书中曾说过:“不可重复的单一事件对科学没有意义”(1959年)。独立重复实验和分析是我们评估科学发现有效性的黄金标准。不幸的是,大多数测序实验的成本太高,无法从试管中重复,因此我们越来越只依赖于生物信息学的再现性。生物信息学项目的复杂性通常会阻碍复制,因此,作为一名优秀的科学家,我们的工作是通过使其更容易地促进和鼓励生物信息学的再现性。正如我们稍后将看到的,采用良好的重现性实践也可以使您作为一名研究人员的生活更轻松。
那么什么是可复制的生物信息学项目呢?至少,它要求共享项目的代码和数据。大多数期刊和资助机构都要求你共享你的项目数据,而像NCBI的SRA数据库这样的资源就是为了这个目的而存在的。现在,编辑和审查人员通常会建议(或者在某些情况下需要)项目的代码也要共享,特别是如果代码是研究结果的重要组成部分。然而,为了确保我们的项目的可再现性,我们可以也应该做很多事情。通过重复生物信息学分析来验证结果,我从中了解到很多让人头疼的问题往往在细节中。
例如,我和同事曾经有一段时间很难重复我们自己做的转录组差异表达分析。几周前,我们对一部分样本进行了分析,得到了初步结果,但令我们惊讶的是,我们目前的分析结果产生了一组非常小的差异表达基因。在重新检查了我们过去的结果是如何创建的,比较了数据版本和文件创建时间,并查看了分析代码的差异之后,我们仍然感到困惑,没有什么能解释结果之间的差异。最后,我们检查了R包的版本,发现它已经在我们的服务器上更新了。然后我们重新安装了旧版本,以确认这是造成差异的原因,事实上确实如此。这里的教训是,无论您将来还是其他人,重复通常不仅依赖数据和代码,还依赖软件版本、数据下载时间和版本等详细信息。这些元数据或数据是确保再现性的关键细节。
另一个激发生物信息学可重复性的案例研究是所谓的“杜克传奇”。杜克大学的Anil Potti博士和其他研究人员创造了一种方法,利用高通量微阵列的表达数据来检测和预测对不同化疗药物的反应。这些方法是个性化药物新水平的开始,并被用于临床试验中确定患者的化疗治疗。然而,两位生物统计学家Keith Baggerly和Kevin Coombes在试图复制本研究时发现了严重的缺陷(Baggerly和Coombes,2009年)。其中许多都需要巴格利和库姆斯所说的“法医生物信息学”,即当没有足够的文件来追溯每一步时,对研究结果进行调查,试图重现研究结果。巴格利和库姆斯总共发现了多个严重错误,包括:
- 一个“差一错误”,导致所有基因对应的表达量都发生了错位。
- 两个具有生物学意义的基因数据被视为“离群值”而被丢弃。
- 进行测序的时间有错。
- 样品分组命名混淆。
Baggerly和Coombes的工作最好在他们的开放访问文章“从细胞系衍生化学敏感性:法医生物信息学和高通量生物学中的可重复研究”中进行总结(参见本章的Github目录了解本文以及杜克传奇的更多信息)。巴格利和库姆斯的工作的教训是,“常见的错误很简单,简单的错误很常见”,糟糕的文档会导致错误和不可生产性。方法、数据版本和代码的文档记录不仅有助于再现性,而且很可能在他们的研究中避免了一些严重的错误。在你的项目中争取最大的再现性常常会使你的工作更加稳健。
稳健的研究和生物信息学的黄金法则
计算机就像一把双刃剑
— The Technical Skills of Statistics
(1964) John Tukey 在湿实验中,当实验失败时,它可能非常明显,但在计算中并不总是如此。如果电泳凝胶看起来像罗夏斑点,而不是整齐的条带,表明显然出了问题。在科学计算中,错误可以是无声的;也就是说,代码和程序可能产生输出(而不是错误地停止),但这个输出可能是错误的。当你学习生物信息学时,这是一个非常重要的概念。
此外,在科学计算中,代码只运行一次是很常见的,因为研究人员得到了他们想要的输出,并进入下一步。相比之下,考虑一个电子游戏:它运行在数千台(如果不是数百万台)不同的机器上,实际上,它经常被许多用户测试。如果出现了删除用户分数的错误,用户很可能会很快发现并报告错误。不幸的是,大多数生物信息学项目并非如此。
要进行稳健的基因组学数据分析也是一项挑战。首先,大多数生物信息学分析产生的中间输出太大、维度太高,无法检测或易于可视化。大多数分析也涉及多个步骤,因此即使检查整个中间数据集是否存在问题是可行的,也很难对每个步骤进行检查(因此,我们通常采用检查数据样本或查看数据汇总统计)。第二,在湿实验室生物学中,通常更容易对实验结果形成预先的预期。例如,研究人员可能期望在某些组织中看到一个特定的mRNA表达,其丰度比一个特定的看家基因要低。有了这些先前的期望,一个异常的结果可以归因于失败的分析而不是生物学。相比之下,大多数基因组学结果的高维度使得几乎不可能形成强烈的先验期望。通过RNA-seq实验对成千上万个基因的表达形成特定的先验期望是不现实的。不幸的是,如果没有事先的预期,很难区分好的结果和坏的结果。
生物信息学家也必须小心,生物信息学项目,甚至是经过大型用户群测试的工具,如比对工具和组装工具,也可能无法在特定有机体上很好地工作。有机体之间的差异有时候大得惊人,它们的基因组也是如此。许多生物信息学工具在一些模式的二倍体生物(如人类)上进行了测试,而在生命树其他部分的复杂基因组上的测试则不那么好。我们真的期望从一个调整到人类数据的短读段比对工具获得的开箱即用的参数能在一个四倍体基因组上适用吗?大概不会。
确保一切正常工作的简单方法是采取谨慎的态度,检查计算步骤之间的一切。此外,您应该以健康的怀疑态度来处理生物学数据(无论是来自实验还是来自数据库),即它可能有问题。在计算社区中,这与“垃圾输入,垃圾输出”的概念有关——分析只和输入数据一样好。在生物信息学教学中,我经常把这一观点作为生物信息学的黄金法则:
别相信你的工具(或数据) 这并不是说生物信息学没有一个值得信任,或者你必须测试你的数据上所有可用的程序和参数。相反,这是训练你采取同样谨慎的态度,软件工程师和生物信息学家已经学会了艰难的方法。简单地检查输入数据和中间结果,运行快速的健全性检查,维护适当的控制和测试程序是一个很好的开始。这还可以避免以后遇到错误,因为修复错误意味着要重做大量的工作。正如你自然而然会测试实验室技术是否有效并得到一致的结果,稳健的生物信息学方法只是在做同样的事情。
形成稳健和可重复的习惯也会让你的生活过得更容易
在科学界工作已经教会了我们许多人生活中的一些事实。这就像墨菲定律:任何可能出错的事情一定会出错。生物信息学有自己的一套这样的规律。在现场工作并与其他生物信息学家讨论过战争故事之后,我可以实事求是地保证以下情况会发生:
几乎可以肯定,您必须多次重新运行分析,可能需要使用新的或更改的数据。经常会发生这种情况,因为你会发现一个bug,合作者会添加或更新一个文件,或者你想在步骤的上游尝试一些新的东西。在所有情况下,下游分析都依赖于这些早期结果,这意味着需要重新运行分析的所有步骤。
在未来,你(或你的合作者,或顾问)几乎肯定需要重新访问项目的一部分,它看起来完全是神秘的。你唯一的辩护就是记录每一步。如果不写下关键的事实(例如,从何处下载数据、下载数据时以及所执行的步骤),您肯定会忘记它们。记录你的计算工作就相当于把一本详细的实验笔记本作为科学的一个绝对重要的部分。
幸运的是,养成能够使您的项目具有可重复性的习惯也有助于解决这些问题。从这个意义上讲,生物信息学(和一般的科学计算)中的良好习惯既使生活更容易,也会造就可重复的项目。原因很简单:如果项目的每一步都被设计成要重新运行(可能有不同的数据),并且有很好的文档记录,那么它已经可以很好地进行重复了。
例如,如果我们使用脚本自动执行任务,并跟踪所有输入数据和软件版本,那么一键就可以重新运行分析。重复此脚本中的所有步骤要容易得多,因为编写良好的脚本自然会记录工作流(我们将在第12章中对此进行详细讨论)。这种方法还可以节省您的时间:如果您收到新的或更新的数据,您所需要做的就是用新的输入文件重新运行脚本。这在实践中并不难做到;脚本不难编写,计算机擅长执行脚本中列举的重复任务。
进行稳健的研究的建议
稳健的研究主要是采用一系列的习惯,这些习惯将有利于您的沉默错误不会混淆您的结果。如前所述,除了防止可怕的沉默错误之外,这些习惯在很多方面都是有益的,这是采纳以下建议的更重要的理由。
关注你的实验设计
稳健的研究始于良好的实验设计。不幸的是,没有多少出色的分析可以挽救一个设计糟糕的实验。引用一位杰出的统计学家和遗传学家的话:
实验结束后咨询统计学家通常只是要求他进行尸检。他也许能说出实验的死亡原因。
—R.A. Fisher 这句话切中要害;我已经看到项目落在我的办公桌上准备分析,在花费了数千美元的测序之后,但它们在到达时已经完全死掉了。好的实验设计并不一定很困难,但因为它从根本上说是一个统计主题,所以它不在本书的范围之内。我之所以提到这个话题,是因为不幸的是,这本书中没有其他东西可以拯救一个设计糟糕的实验。尤其有必要考虑高通量研究中的实验设计,因为技术“批次效应”会显著干扰研究(有关这方面的观点,请参见Leek等人,2010)。
大多数统计学入门课程和书籍涵盖了实验设计的基本主题。Quinn和Keough的《生物学家实验设计和数据分析》(剑桥大学出版社,2002年)是一本关于这一主题的优秀书籍。《O'Reilly's Statistics in a Nutshell》第二版第18章,作者Sarah Boslaugh,也很好地涵盖了基础知识。然而,请注意,基因组学实验中的实验设计是另一种野兽,并且正在被活跃地研究和改进。要确保您花了几千美元做的实验充分发挥作用,最佳方法是看看当前针对您的项目的最佳设计。最好咨询统计学家,了解您在设计实验时可能遇到的任何实验设计问题或关键点。
写代码给人看,写数据给电脑看
调试比一开始编写代码要困难两倍。因此,如果您尽可能巧妙地编写代码,那么根据定义,您就没有足够的智能来调试它。
—Brian Kernighan 生物信息学项目可能涉及大量的代码,而我们防止错误的最佳防御措施之一是为人类而不是为计算机编写代码(Wilson等人,2012年的优秀文章中指出了这一点)。人类是进行调试的人,因此编写简单、清晰的代码使调试更加容易。
代码应该是可读的,可以分解为小的包含组件(模块化)和可重用的(所以您不需要反复地重写代码来完成相同的任务)。这些实践在软件世界中是至关重要的,也应该应用于您的生物信息学工作中。注释代码和采用样式指南是提高代码可读性的简单方法。谷歌有多种语言的公共风格指南,它们是很好的模板。为什么代码可读性如此重要?首先,可读代码使项目更具可重复性,因为其他人更容易理解脚本的作用和工作方式。第二,在可读、注释性好的代码中查找和纠正软件错误要比混乱的代码容易得多。第三,当代码被很好地注释和清晰地编写时,在将来重新访问代码总是容易的。编写模块化的和可重用的代码需要实践,我们将在本书中看到这方面的一些例子。
与代码相比,数据的格式应便于计算机可读。通常,我们作为人类记录数据的方式可以最大限度地提高其可读性,但在计算机处理数据之前,需要进行大量的清理和整理。计算机可读的数据(和元数据)越多,我们就越能利用计算机处理这些数据。
让你的电脑帮你干活
做机械性的事情的人往往会犯很多错误。使你的工作更稳健的最简单的方法之一就是让你的计算机尽可能地做这些机械性的工作。这种自动化任务的方法更为稳健,因为它减少了犯一个小错误的可能性,例如意外地忽略了一个文件或错误地命名输出。
例如,通过单独键入(或复制和粘贴)在20个不同的文件上运行一个程序,每个命令都是脆弱的,随着处理的每个文件,犯错误的几率会增加。在生物信息学工作中,养成让计算机为你做这种重复工作的习惯是很好的。不要将同一个命令粘贴20次,只更改输入和输出文件,而是编写一个为您执行此操作的脚本。这不仅容易而且不太可能导致错误,而且还增加了可重复性,因为您的脚本可以作为您对每个文件所做操作的参考。
利用自动化任务的好处需要在组织项目、数据和代码时考虑一下。简单的习惯,如以计算机(而不仅仅是人类)能够理解的一致方式命名数据文件,可以极大地促进任务的自动化,使工作更加容易。我们将在第2章中看到这方面的例子。
在代码和方法中做出响亮的断言
当我们编写代码时,我们倾向于对数据有隐含的假设。例如,我们期望只有三种DNA链选择(正向、反向和未知),一个基因的起始位置小于结束位置,我们不能有负的位置。我们对数据所做的这些隐式假设会影响我们编写代码的方式;例如,如果我们假设代码不会发生,我们可能不会考虑在代码中处理某些情况。不幸的是,这会导致可怕的无声错误:我们的代码或程序接收到超出预期值的值,行为不正确,但仍然返回输出而不发出警告。防止此类错误的最佳方法是使用诸如python的assert()和r的stopinfot()等assert语句显式地声明和测试我们对代码中数据的假设。
几乎每种编程语言都有自己版本的断言函数。这些断言函数的操作方式类似:如果语句的计算结果为false,则断言函数将停止程序并引发错误。它们可能很简单,但这些断言函数在健壮的研究中是必不可少的。在我职业生涯的早期,一位导师激励我养成使用断言的习惯,即使这种说法似乎绝对不可能是错误的,但我不断地感到惊讶的是,有多少次,这些声明中出现了微妙的错误。在生物信息学(以及所有领域)中,我们必须尽可能地使程序可以将可怕的无声错误报告出来。
测试代码,如果可以的话,让代码测试代码
软件工程师是一群聪明的人,他们把让计算机把工作提升到新的水平的想法付诸实践。一种方法是让代码测试其他代码,而不是手工进行。测试代码的一种常见方法称为单元测试。在单元测试中,我们将代码分解为单独的模块单元(这也有提高可读性的副作用),并编写额外的代码来测试此代码。实际上,这意味着如果我们有一个名为add()的函数,我们会编写一个名为test_add()的附加函数(通常在单独的文件中)。这个test_add()函数将调用带有特定输入的add()函数,并测试输出是否如预期的那样。在python中,这可能看起来像:
EPS = 0.00001 # a small number to use when comparing floating-point values
def add(x, y):
"""Add two things together."""
return x + y
def test_add():
"""Test that the add() function works for a variety of numeric types."""
assert(add(2, 3) == 5)
assert(add(-2, 3) == 1)
assert(add(-1, -1) == -2)
assert(abs(add(2.4, 0.1) - 2.5) < EPS)测试函数的最后一行看起来比其他行复杂,因为它比较的是浮点值。很难在计算机上比较浮点值,因为存在表示错误和舍入错误。然而,这是一个很好的提醒,我们总是被机器的功能所限制,我们必须在分析中注意这些限制。
尽管科学代码更有可能包含错误(因为我们的代码通常只运行一次以生成出版物的结果,而且科学代码中的许多错误都是无声的),但与软件行业相比,在科学编码中使用的单元测试要少得多。我把这称为科学编码的悖论:科学编码的易出错性意味着我们应该尽可能多地利用测试,但实际上我们所做的测试(如果有的话)要少得多。这是令人遗憾的,因为现在许多科学结论都是堆积如山的代码的结果。
虽然测试代码是发现、修复和防止软件错误的最佳方法,但是测试并不便宜。测试代码使我们的结果更加稳健,但也需要花费大量的时间。不幸的是,对于研究人员来说,为他们编写的每一段代码编写单元测试需要花费太多的时间。科学发展很快,在编写和执行单元测试所需的时间内,您的研究可能会过时或被抢走。更明智的策略是,每次编写代码时考虑三个重要变量:
- 这些代码被其他代码调用的频率多高?
- 如果这段代码有误,对最终结果会造成多大的影响?
- 如果出现了错误,这个错误有多显而易见?
测试代码的重要性与前两个变量成正比,与第三个变量成反比(也就是说,如果一个bug非常明显,那么就没有必要为它编写测试)。我们将在本书的整个例子中采用这种策略。
尽量用现成的函数库
每一个初学程序员的职业生涯都有一个转折点,当他们觉得编写代码足够舒服时,他们会想,“嘿,为什么我要使用一个库,我自己就可以轻松地编写这个库。”这是一种让自己充满力量的感觉,但使用现有的软件库是有很好的理由的。
与您自己编写的库相比,现有的开放源码库有两个优势:历史更长,读者更广泛。这两个优点都可以减少错误。软件中的错误类似于众所周知的在大海捞针的问题。如果你编写自己的软件库(其中一定会有一些bug潜藏),你就是一个人在找一些针。相比之下,开放源码软件库在本质上有更多的人用更长的时间在寻找这些针。因此,在这些开放源码库中发现、报告和修复bug的可能性要高于您自己的自制版本。
一个很好的例子是,当编写脚本将核苷酸转化为蛋白质时,可能会出现一个微妙的问题。大多数有编程经验的生物学家可以很容易地编写一个脚本来完成这项任务。但这些简单的编程练习背后隐藏着隐藏的复杂性,你可能不会考虑。如果你的核苷酸序列中有ns怎么办?或者Ys?或者Ws?Ns、Ys和Ws可能看起来不是有效的碱基,但它们是国际纯化学和应用化学联合会(IUPAC)标准的简并核苷酸,完全适用于生物信息学。在许多情况下,经过良好审查的软件库已经发现并修复了这些隐藏的问题。
把数据看成是只读的
许多科学家花费大量时间使用Excel,不用一眨眼的时间,就会改变单元格内的值并保存结果。我强烈建议不要这样修改数据。相反,更好的方法是将所有数据视为只读,只允许程序读取数据并创建新的、独立的结果文件。
为什么在生物信息学中将数据视为只读的很重要?首先,就地修改数据很容易导致结果损坏。例如,假设您编写了一个直接修改文件的脚本。在处理一个大文件的过程中,你的脚本报错并中断了。由于你修改了原始文件,你又没办法撤销这个修改,最终导致你的原始文件不能再用了。
其次,当我们修改一个文件时,很容易忘记我们是如何更改它的。与每个步骤都有一个输入文件和一个输出文件的工作流不同,一个就地修改的文件不会给我们任何关于我们对它做了什么的指示。如果我们忘记了如何更改文件,并且没有原始数据的备份副本,那么我们的更改基本上是不可复制的。
对于熟悉Excel的科学家来说,将数据视为只读似乎有悖常理,但这对于稳健的研究至关重要(可以防止灾难,并有助于再现性)。最初的困难是值得的;除了保护数据不受损坏和不正确的更改之外,它还促进了再现性。此外,分析的任何步骤都可以很容易地重做,因为输入数据是由程序改变的。
把常用的脚本整理成工具
不断地确保你的数据是没问题的
如何使研究可重复
采用可重复的研究方法并不需要花费太多的精力。和稳健的研究习惯一样,可重复的方法最终会使你的生活更容易,因为你可能需要在你忘记了细节很久之后再重复你过去的工作。以下是在实践生物信息学以使您的工作具有可复制性时需要考虑的一些基本建议。
发布你的数据和代码
对于可重复性,绝对的最低要求是释放代码和数据。如果没有可用的代码和数据,您的研究是不可复制的(参见Peng,2001)。稍后我们将在本书中讨论如何共享代码和数据。
记录所有东西
科学家走进实验室的第一天,他们被告知要保存一本实验室记录本。可悲的是,这种良好的习惯常常被计算领域的研究人员所抛弃。发布代码和数据是对可重复性的最低要求,但是大量的文档也是再现性的重要组成部分。要完全重现一项研究,每一个分析步骤都必须比学术文章中描述的更详细。因此,附加文件对于可重复性是必不可少的。
要采用的一个好做法是将每个分析步骤记录在纯文本README文件中。像一本详细的实验记录本一样,这个文档可以作为您步骤的宝贵记录,记录文件的位置、来源或包含的内容。这个文档可以与项目的代码和数据一起存储(我们将在第2章和第5章中看到更多关于这个的内容),这可以帮助合作者了解您所做的工作。文档还应该包括每个执行的程序的所有输入参数、这些程序的版本以及它们的运行方式。现代软件如R的 Knitr和iPython Notebooks是记录研究的强大工具;我在本章的GitHub README中列出了一些开始使用这些工具的资源。
把脚本的运行结果制作成图像或者统计结果
要确保一个科学项目是可重复的,不仅涉及对关键统计检验的可重复性,支持科学发现的要素(如图表)也应是可重复的。确保这些要素可重复的最佳方法是让每个图像或表都成为脚本(或脚本)的输出。
编写脚本以生成图像和表格似乎比在Excel或R中交互生成这些图像和表格更耗时。但是,如果在更改某个步骤后必须手动重新生成多个图形,那您就能知道这种方法的优点。生成表和图像的脚本可以很容易地重新运行,节省您的时间,并使您的研究更具可重复性。像iPython Notebook和knitr(在前一节中提到)这样的工具也可以对完成这些任务起极大的帮助。
用代码来写文档
对于复杂的处理流程,最好的文档通常是被良好注释的代码。因为代码足以告诉计算机如何执行一个程序(以及要使用哪些参数),它也足以告诉人类如何重复您的工作(软件版本和输入数据等附加信息也是完全可重复的)。在许多情况下,编写一个脚本来执行分析的关键步骤比输入命令然后将它们记录到其他地方更容易。同样,代码是一件美妙的事情,使用代码来记录分析的每一步,这意味着如果需要,可以很容易地重新运行分析的所有步骤——只需重新运行脚本即可。
不断地提高你的生物信息数据技能
在你完成本书的其余部分工作时,将本章介绍的基本思想放在心中。我在这里介绍的内容足以让您开始思考稳健和可重复的生物信息学中的一些核心概念。这些主题中的许多(例如,可重复性和软件测试)目前仍在积极研究中,我鼓励感兴趣的读者更深入地研究这些主题(我在本章的Github README中包含了一些资源)。
原文地址:https://zhuanlan.zhihu.com/p/351414526 |
|

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 






 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 发表于 2025-5-10 06:22
发表于 2025-5-10 06:22



