只需一步,快速开始
微信扫一扫,快速登录
您需要 登录 才可以下载或查看,没有账号?立即注册
1.开展临床试验,应当获得药品监督管理部门许可,生物等效性试验应按照要求完成备案。
2.具有药物临床试验伦理委员会批件。
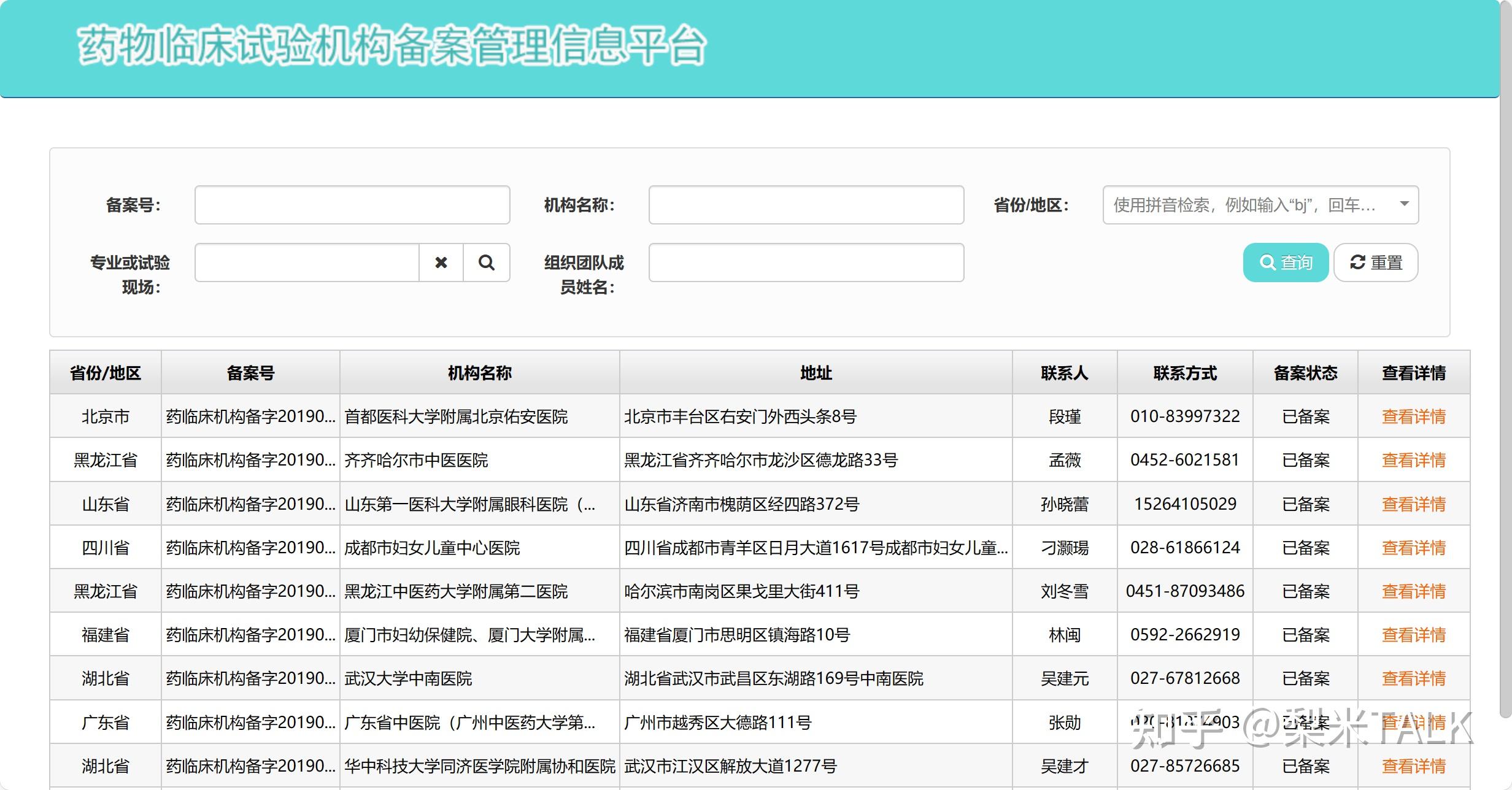
3.药物临床试验在具备相应条件并按规定备案的药物临床试验机构(以下简称“临床试验机构”)开展。其中,疫苗临床试验由符合国家药品监督管理局和国家卫生健康委员会规定条件的三级医疗机构或者省级以上疾病预防控制机构实施或者组织实施。
4.临床试验实际开展场地与申报资料中试验地址一致,具备临床试验所需设施设备,检定、校准和日常维护符合要求,医疗急救设施保证有效运转。
5.临床试验机构及专业制定与工作相适应的管理文件,并遵照执行。管理文件符合法规及指导原则等的要求,能够覆盖临床试验的全过程。
6.临床试验各环节参与人员具有能够承担临床试验工作相应的教育、培训和经验,并得到主要研究者的授权。
7.研究者、临床试验机构与申办者在试验开始前签署临床试验合同,对相关的权利与义务进行约定。
8.申办者/合同研究组织(CRO)按照药物临床试验质量管理规范(GCP)、临床试验方案及合同履行了相应职责,并保存相关文件和记录。
9.医疗机构临床实验室保证检验检测系统的完整性和有效性,对需要校准的检验仪器、对临床检验结果有影响的辅助设备及临床试验需要的其他设备等进行定期校准。
10.医疗机构临床实验室参加经国家卫生健康部门认定的室间质量评价机构组织的临床检验室间质量评价并取得通过证书。
举报
本版积分规则 发表回复 回帖后跳转到最后一页
查看 »
微信扫一扫关注本站公众号

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 






 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 发表于 2025-3-13 22:59
发表于 2025-3-13 22:59