金桔
金币
威望
贡献
回帖0
精华
在线时间 小时
|
登陆有奖并可浏览互动!
您需要 登录 才可以下载或查看,没有账号?立即注册


×
2005年,罗氏推出了第一款二代测序仪罗氏454,生命科学开始进入高通量测序时代。后续随着Illumina系列测序平台的推出,极大降低了二代测序的价格,推动了高通量测序在生命科学各个研究领域的普及。目前,高通量测序已经成为一种常规研究方法,大量科研工作中均会用到。然而,为什么二代测序能实现高通量?为什么二代测序读长如此之短?为什么reads末端测序质量会降低?应该如何选择测序读长与打断片段的长度?想要回答这些问题,都需要详细了解二代测序的基本原理。本篇文章以典型的Illumina双末端测序为例,详细解析二代测序的原理。
第二代测序(Next-generation sequencing,NGS)又称为高通量测序(High-throughput sequencing),是基于PCR和基因芯片发展而来的DNA测序技术。我们都知道一代测序为合成终止测序,而二代测序开创性的引入了可逆终止末端,从而实现边合成边测序(Sequencing by Synthesis)。二代测序在DNA复制过程中通过捕捉新添加的碱基所携带的特殊标记(一般为荧光分子标记)来确定DNA的序列,现有的技术平台主要包括Roche的454 FLX、Illumina的Miseq/Hiseq等。由于在二代测序中,单个DNA分子必须扩增成由相同DNA组成的基因簇,然后进行同步复制,来增强荧光信号强度从而读出DNA序列;而随着读长增长,基因簇复制的协同性降低,导致碱基测序质量下降,这严格限制了二代测序的读长(不超过500bp),因此,二代测序具有通量高、读长短的特点。二代测序适合扩增子测序(例如16S、18S、ITS的可变区),而基因组、宏基因组DNA则需要使用鸟枪法(Shotgun method)打断成小片段,测序完毕后再使用生物信息学方法进行拼接。
原文地址(关注公众号微生态与微进化领取原理讲解视频):
文库构建
文库构建即为测序片段添加接头。无论是PCR产生的片段还是基因组鸟枪法打断的片段都具有特异性(PCR中不同样品反向引物插入了特异性的barcode,因此两端也是特异的),两端缺乏必要的引物因此混合DNA片段不能直接扩增和测序。DNA片段需要加接头修饰才能进行上机测序,这个过程称为二代测序的文库构建。下面我们以常用的试剂盒NEBNext®Ultra™ II DNA Library Prep Kit for Illumina®为例阐述二代测序文库构建的流程及其原理,具体如下所示:
①末端修饰。目前很多PCR使用的高保真Pfu聚合酶产生的片段末端是平齐的(也即没有不配对的碱基);鸟枪法产生的片段则是随机断裂,其末端可能是平齐的也可能是不平的。因此,建库第一步是使用Taq聚合酶补齐不平的末端,并在两个末端添加突出的碱基A,从而产生粘性末端(若使用Taq酶扩增,则无需末端修饰),产生粘性末端的片段可以添加接头(Adaptor)。
②添加接头。经过末端修饰后的PCR片段末端具有突出的A尾,而接头具有突出的T尾,可以使用连接酶将接头添加到DNA片段两端。NEB的接头为特殊的碱基U连接的环状结构(可以增强稳定性),因此连接接头后,还需要将碱基U删除从而形成“Y”形接头。这一步添加的接头主要是为了后续PCR中作为引物扩增继续添加文库index和与测序平台互补的寡核苷酸序列(此外还作为测序引物Rd1 SP/Rd2 SP),而之所以为“Y”型开叉结构,是因为每一端接头是两条不互补的序列(每一端都是Rd1 SP与Rd2 SP交错),因为连接酶没有选择性,每个接头都是只靠突出的T来与DNA连接,“Y”接头保证了每条单序列两端均为不同的测序引物,从而在后续PCR中可以连接不同的寡核苷酸序列(P5/P7),具体流程见下图。
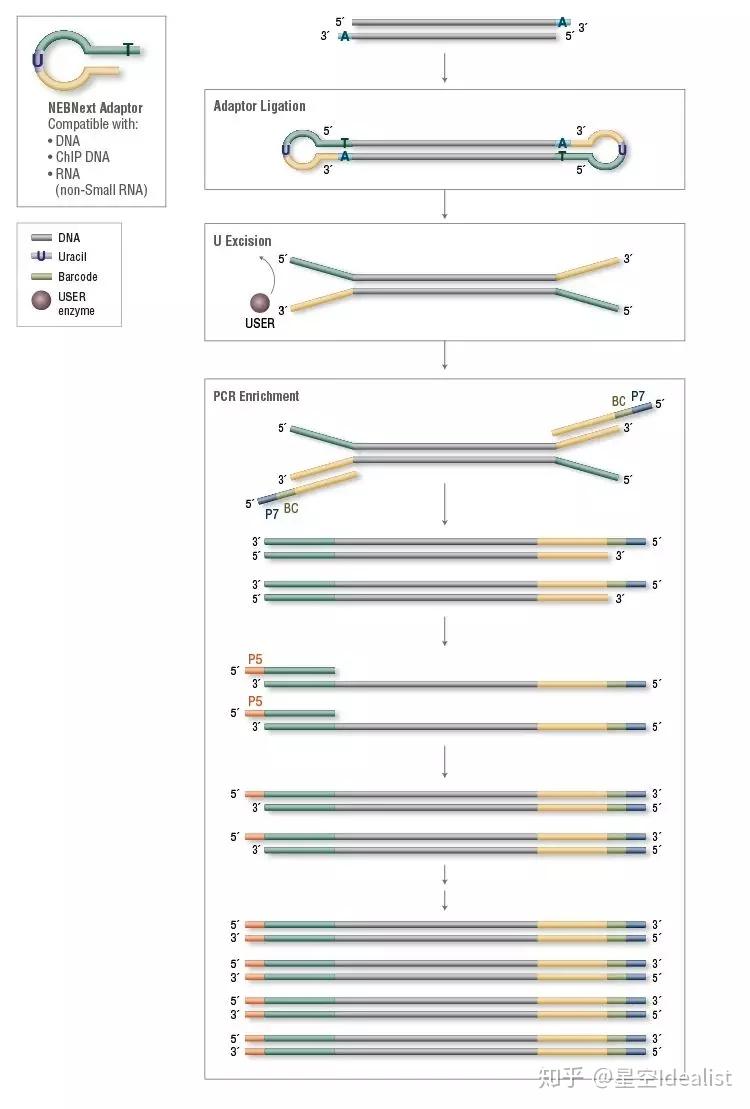
③磁珠纯化。添加接头后的文库体系中含有聚合酶、连接酶等各种酶以及辅助物质,接头的添加也是过量的,而且由于末端的不稳定性,容易形成自连片段,鸟枪法打断的片段中也可能有大片段存在,所以需要特殊磁珠(AMPure XP Beads)纯化来去除大片段以及各种杂质,从而获得成功添加接头的文库片段。其原理为磁珠可以通过氢键等作用力来吸附DNA片段,磁珠本身不具有片段大小选择的能力,但其储存的buffer里面含有20%的PEG 8000,PEG浓度越大则可以吸附的DNA片段越小。因此磁珠纯化的时候要根据文库片段不同严格控制磁珠添加量(其实是PEG添加量)来实现片段选择。
④PCR扩增。添加了接头的DNA片段,可以使用与接头互补的引物来扩增。这个过程非常重要,因为目前所有片段其两端是不互补的Y形结构,不能直接进行测序;此外,片段还需要添加用于区分不同文库的特异性index,以及与测序仪芯片互补的两种寡核苷酸序列(P5/P7)。
⑤第二次磁珠纯化。PCR后需要将产物DNA片段与聚合酶等杂质分离,因此再次进行磁珠纯化,之后进行质量检测,包括DNA浓度检测、琼脂糖凝胶电泳和片段长度检测,完成建库。
测序是以单链为单位的,建库完成后的每条DNA的单链均一端连有测序引物Read1 Sequencing Primer(Rd1SP)和P5,另一端为Rd2 SP、Index(Barcode)和P7。Index用来区分不同的文库,因为测序仪一个run产生数据量巨大,由于实际情况不同,一次上机常会进行多个文库测序,因此需要加上Index来区分。
经评论区指正这里做补充说明:
在建库过程中,文库中每个DNA短片段的正链与反链都加上了P5与P7,因此建库后每个DNA片段都会扩增出两种结果(详见上面插图),如果全部上机,最终两条链都会有测序结果。因为上机测序起始是以DNA单链为单位,单链化的DNA片段进入测序仪流通池,会随机的结合在不同位置,且相互距离足够远以保证测序信号的独立读取。最终获得的测序结果会有重复的reads(反向互补也会有重复),所以都会有去重步骤,而且测序量越大重复率会越高。
上机测序
Illumina测序技术为基于基因芯片的边合成边测序,整个平台可解剖为三个系统:一温度控制系统,原理和普通PCR仪一样,来控制反应的进行;二酶控制系统,通过各种酶来控制DNA合成与剪切;三荧光信号收集系统,可以理解为分辨率极高的照相机。在Illumina测序平台的流通池(Flow cell)表面,通过基因芯片技术交错固定了无数条寡核苷酸链(即短核苷酸链),分别为P5'(P5互补)和P7,单链化的文库DNA片段进入流通池后,包含P5或P7'的单链可以与表面的寡核苷酸基于互补配对结合,从而进入测序过程。测序具体流程如下:
①首先以寡核苷酸为引物、文库片段为模板进行DNA复制(因为文库稀释后浓度足够低,可以认为文库片段均匀的结合在流通池表面,每个片段结合的位置相距足够远,这很重要,否则测序时会导致信号叠加而不能识别)。复制完成后解链,将文库片段洗去,留在流通池表面的为与文库模板互补的DNA链。

②因为单链DNA另一端为不同的接头序列,可以与相邻的另一种寡核苷酸互补结合,之后进行“桥”式扩增(假如第一次结合的为P7,则复制完成洗脱模板后顶端可以与相邻的P5互补结合形成“桥”,并以P5为引物进行复制,完成后再次解链并与相邻不同种接头结合来进行复制,如此类推)。25-28个循环完成后,原来散布在表面的单核苷酸序列变成散布的DNA簇,这一步主要是为后续测序做准备,因为测序时单分子产生的光信号很弱,难以检测。
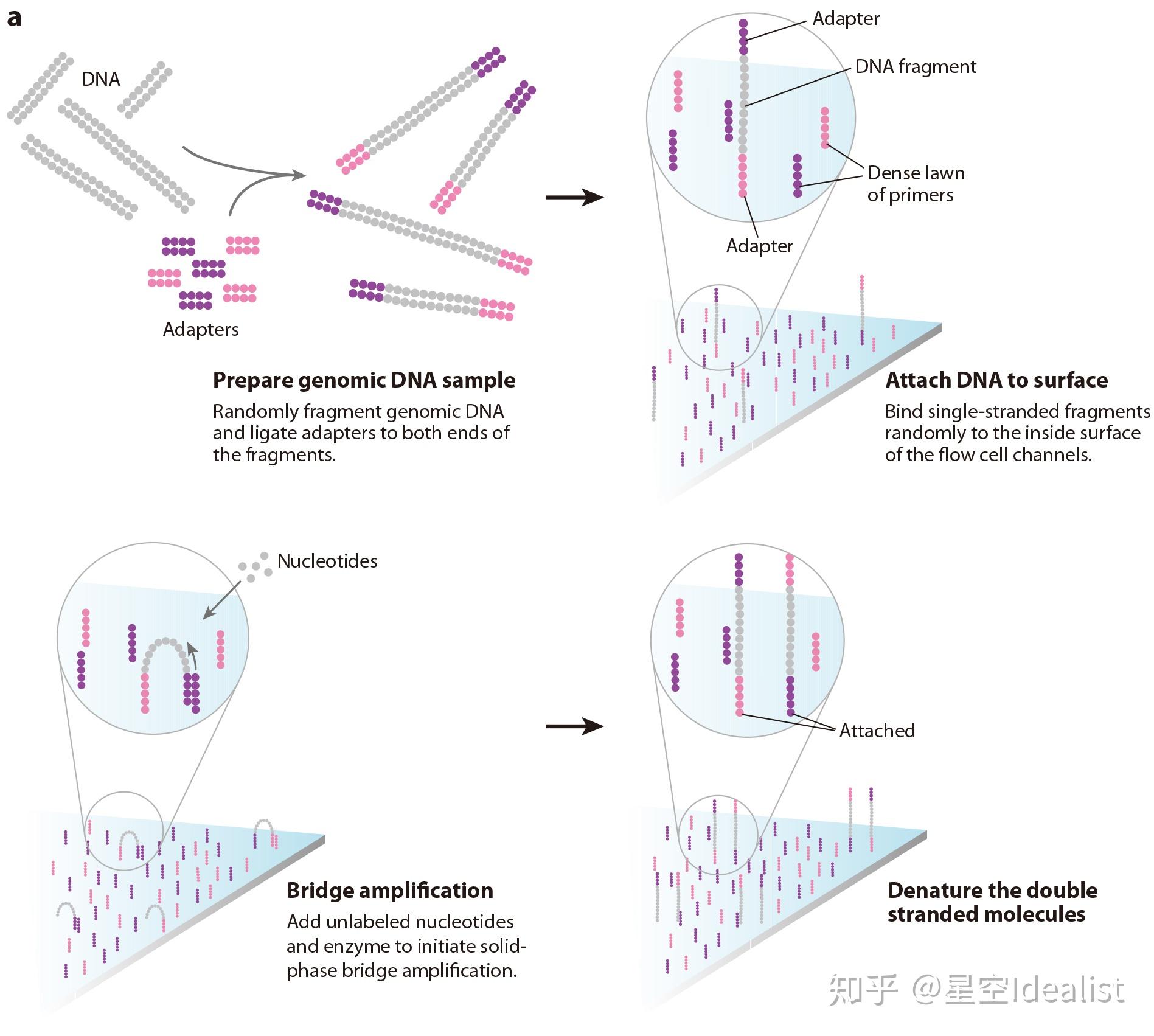
③“桥”式扩增后一个DNA簇都是由最初的一个文库模板复制而来,但是这时候P7上的序列与P5上的序列是分别从两端开始的,测序要保证每个片段一致性(都是正向或都是反向),因此再次解链线性化,切割并洗去P5上的DNA链,只留P7上的DNA单链。Illumina巧妙地利用了甲酰胺基嘧啶糖苷酶Fpg对8-氧鸟嘌呤糖苷8-oxo-G的选择性切断作用,在合成的引物链上加入了一个8-oxo-G,用Fpg处理,就把带8-oxo-G基团切掉,并把DNA链切断,留下一带不完整糖基的磷酸基。这个磷酸基在接下来的过程中,起到了阻止P5延伸的作用。此后的双末端测序中需要恢复3'-OH,则用脱嘌呤嘧啶内切核酸酶AP-endonuclease把带不完整糖基的那个磷酸基切掉。
④加入测序引物Read1 SP和修饰过的DNA聚合酶,则在测序引物3’端开始DNA复制。在流通池加入可逆终止荧光dNTP,其3'-OH被阻隔(糖基3'连接有叠氮基团,在链延伸时起到了阻止添加下一个dNTP作用,因此在除去阻隔前只能添加一个碱基),4种dNTP在碱基上分别连接有不同颜色的荧光基团(也可以相同颜色荧光标记,但是测序会更慢,每次只能添加一种碱基)。之后洗掉多余的dNTP,使用激光扫描,收集留在流通池表面的荧光信号(如图1-6所示)。用巯基试剂去掉3’位阻断的叠氮基团,用TCEP(Tris(2-carboxyethyl)phosphine,三(2-羧乙基)膦)去掉荧光基团,进入下一个碱基的测序反应。因为每条DNA单链扩增形成的DNA簇均固定在表面,随着反应进行根据相同位置出现的荧光信号情况,就逐渐读出了改位点DNA链的序列。
⑤要保证测序的准确性,需要一个位点DNA簇的每条链同步复制,然而随着反应进行,不同链复制情况会出现差异,因此二代测序读长目前限制在300bp以内。Read1结束后,解链并洗掉测序中已经合成的部分,加入测序引物Index引物(也即Read2 SP互补的寡核苷酸),这时会继续在3’端进行复制,读出接头中Index序列,从而可以确定出每个位点的DNA属于哪个文库。
⑥为了增长测序长度,进行另一个方向测序,也即双末端测序。洗掉前面复制合成的片段,DNA单链继续在流通池表面形成桥式连接,这时要用脱嘌呤嘧啶内切核酸酶处理修复P5的3’-OH末端,加入聚合酶,则在P5末端开始DNA复制。十几个循环后,将P7上的DNA切割并洗掉。Illumina通过在P7核酸链中加入一个U碱基,用USER酶(Uracil Specific Excision Reagent,尿嘧啶链特定切断试剂)来切隔断链。这时只留下P5上的DNA链,与Read中方向相反。加入测序引物Read2 SP,进行另一端的序列读取。
测序数据
一般我们接触到的测序数据为fastq格式的碱基序列,然而早期Illumina平台直接下机数据为bcl格式文件,其储存的是显微拍摄得到的荧光信号信息,如下所示(此图为不同碱基使用相同荧光标记的扫描结果):

将相同区域不同时间拍摄的荧光图片按照时间顺序叠加处理,就可以获得该位点结合的DNA序列的碱基顺序。
参考文献
[1] ClarkeA C, Prost S, Stanton J a L, et al. From cheek swabs to consensus sequences: anA to Z protocol for high-throughput DNA sequencing of complete humanmitochondrial genomes[J]. Bmc Genomics, 2014, 15(1): 1-12.
[2] BowmanS K, Simon M D, Deaton A M, et al. Multiplexed Illumina sequencing librariesfrom picogram quantities of DNA[J]. Bmc Genomics, 2013, 14(1): 135-143.
[3] MardisE R. Next-Generation DNA Sequencing Methods[J]. Annual Review of Genomics &Human Genetics, 2008, 9(9): 387-402
原文地址:https://zhuanlan.zhihu.com/p/91913739 |
|

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 






 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 发表于 2025-3-10 21:28
发表于 2025-3-10 21:28



