金桔
金币
威望
贡献
回帖0
精华
在线时间 小时
|
相信做有机合成的科研人员来说,对核磁都不会陌生,在有机合成中,化合物的表征是必不可少的,最基本表征手段就是核磁,核磁氢谱的解析体现一个人的基本功,以及知识储备。很多人在解析核磁的时候,往往先会用ChemDraw模拟一下出峰位置,然后再与自己的样品核磁对照,也有的用MestReNova来模拟,但是在没有数据库的前提下,模拟也不尽如意。但是高斯通过理论计算的方法,来模拟NMR,就比较精确,今天就为大家带来怎么用高斯模拟NMR。首先介绍一下大体过程。首先声明本方法比较适用于高斯新手,对于高手,就任意了。首先,因为Gaussview建立模型新手可能不熟悉,所以你可以用chemoffice,辅助建立模型:即在chemdraw里面画好分子式,导入chem3D里面,生成gjf格式文件,然后导出gjf名称记为12.gjf。接下来编辑文本,对于新手来说,一定注意空行。(注意,因为在windows下因为内存有限,所以输入格式都是Linux版本的)
1输入计算基组:输入保存路径,%chk=12.chk
2:计算方法# opt freq B3LYP/6-31G* scrf(SMD,solvent=chloroform)NMR
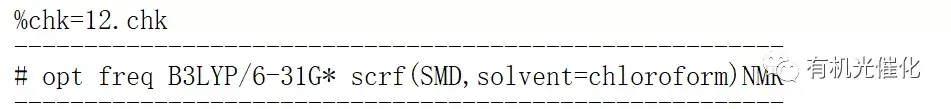
3:Linux计算
在路径下,打开终端,输入:g16 <12.gjf | tee 12.out 回车即可看到计算过程。
假如你需要在Gaussview下观看结果的话,需要将chk 转换成fch才可以在Windows下打开。
具体操作为:formchk 12.chk 回车即可。
量子化学论坛,Sobereva大神的一个帖子说明,标度法是用来计算化学位移的一种又准又便宜的方法,用这种方法计算化学位移(δ)的方式是
δ=(截距-σiso)/(-斜率),(一般用氯仿作溶剂。)截距和斜率通过查表即可得到对应基组的值。(要找到最合用的级别,一方面是看RMSD值(相对于实验值的均方根误差),另一方面是看计算用的基组。RMSD又低,基组又小的级别是我们需要的。)
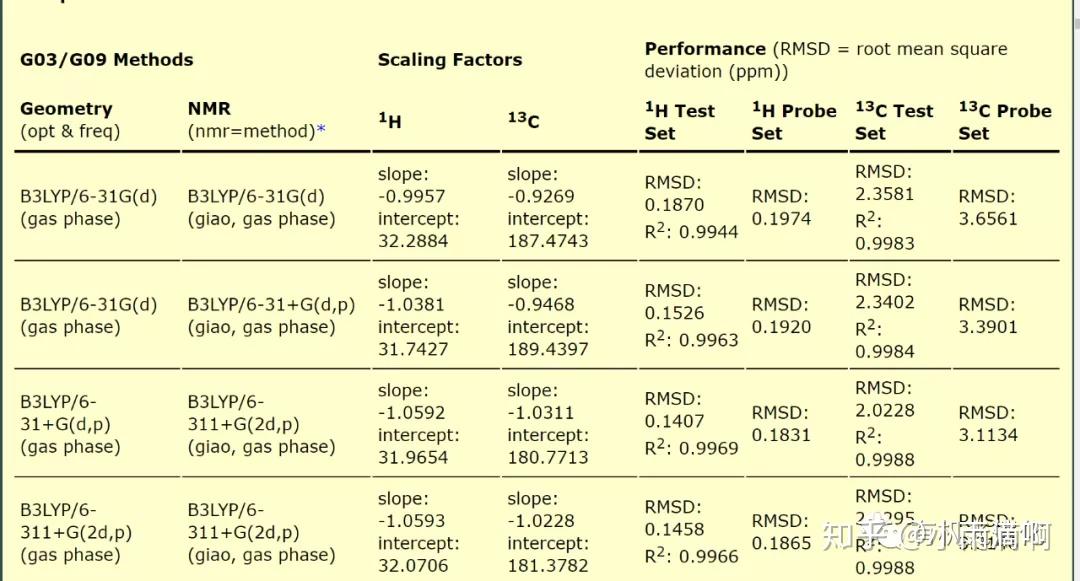
所以我们只需要在计算结果中找到Isotropic(平均值),代入即可。
看不大懂没关系,看看下面两个视频就会啦!
点击下面链接或关注公众号有机光催化,一起进步吧!
核磁模拟谁说了算,高斯告诉你! |
|

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 






 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 发表于 2025-3-10 19:16
发表于 2025-3-10 19:16


 发表于 2025-3-10 19:17
发表于 2025-3-10 19:17



