金桔
金币
威望
贡献
回帖0
精华
在线时间 小时
|
使用MaxQuant解析蛋白质质谱的基本步骤如下:
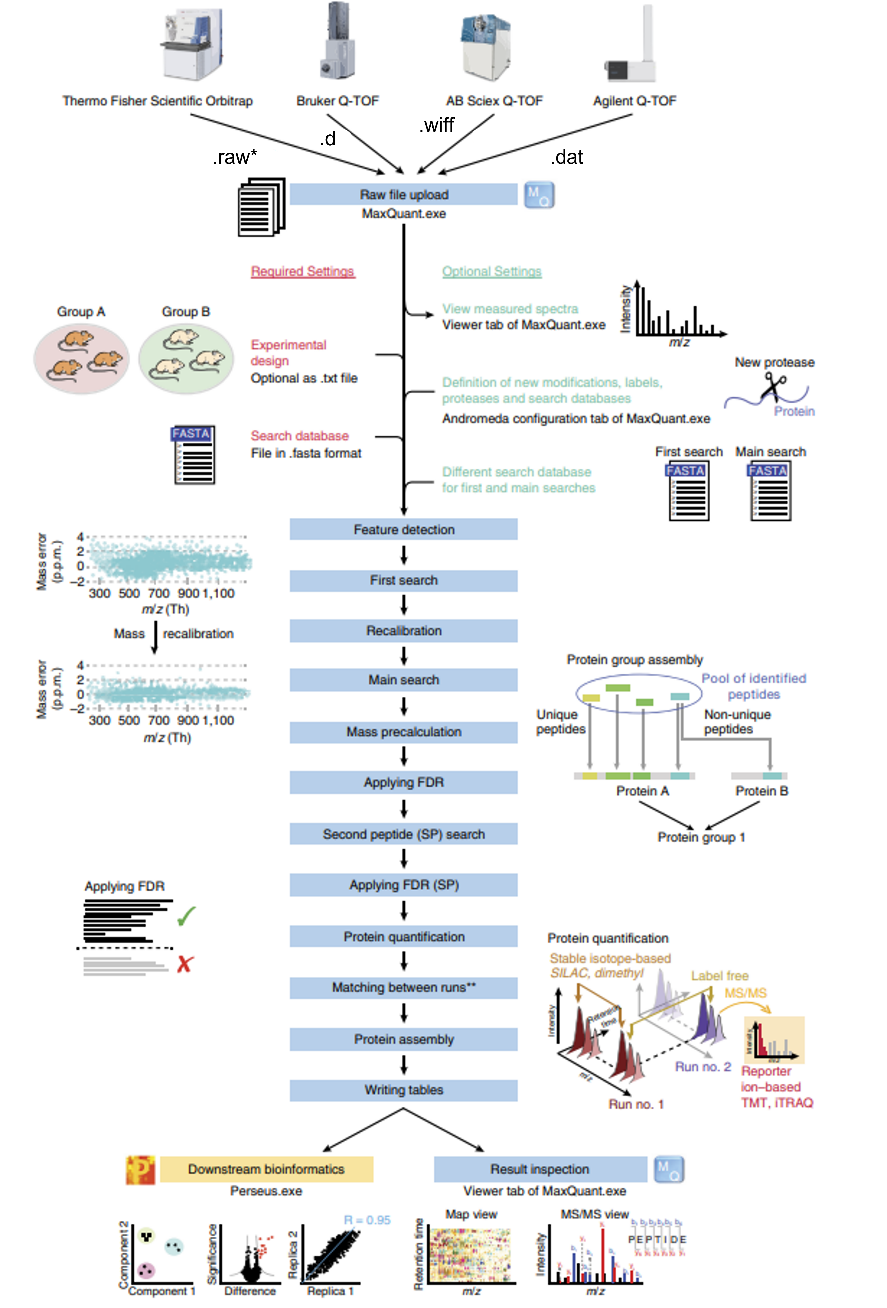
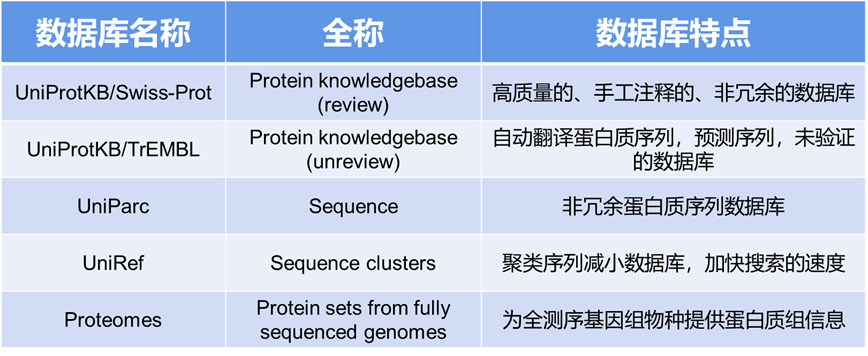
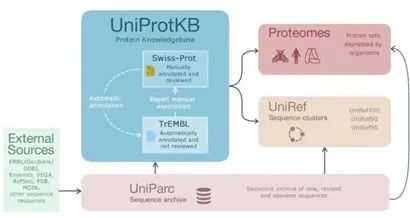
1.数据预处理:收集原始的蛋白质质谱数据,如.raw格式文件,这通常来自质谱设备。需要关于样品的一些基本信息,如蛋白酶的类型(例如,胰蛋白酶)以及修饰。
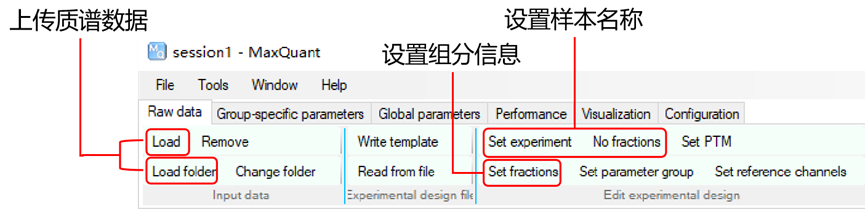
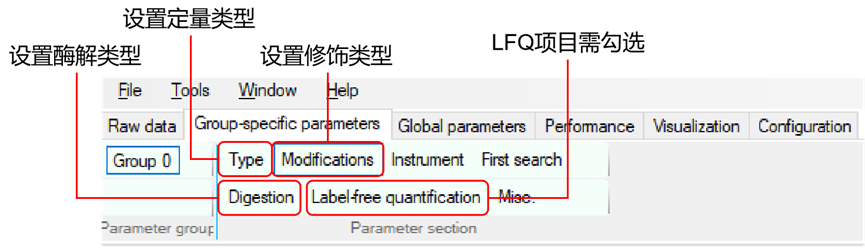
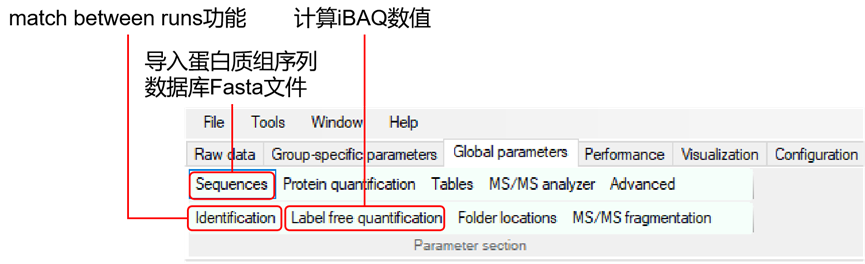
2.设置参数:在MaxQuant中设置您的质谱实验的特定参数。在主界面的左侧,可以看到各种选项卡,如'群组','全局参数', '标准参数'等。在'全局参数'中,需要输入原始数据文件的路径,以及输出文件的路径。在'标准参数'选项卡中,可以设置特定的实验参数,如蛋白酶的类型,修饰等。
3.运行MaxQuant:点击主界面下方的"开始"按钮,MaxQuant会开始分析质谱数据。运行时间可能会有所不同,取决于您的数据的大小和复杂性。
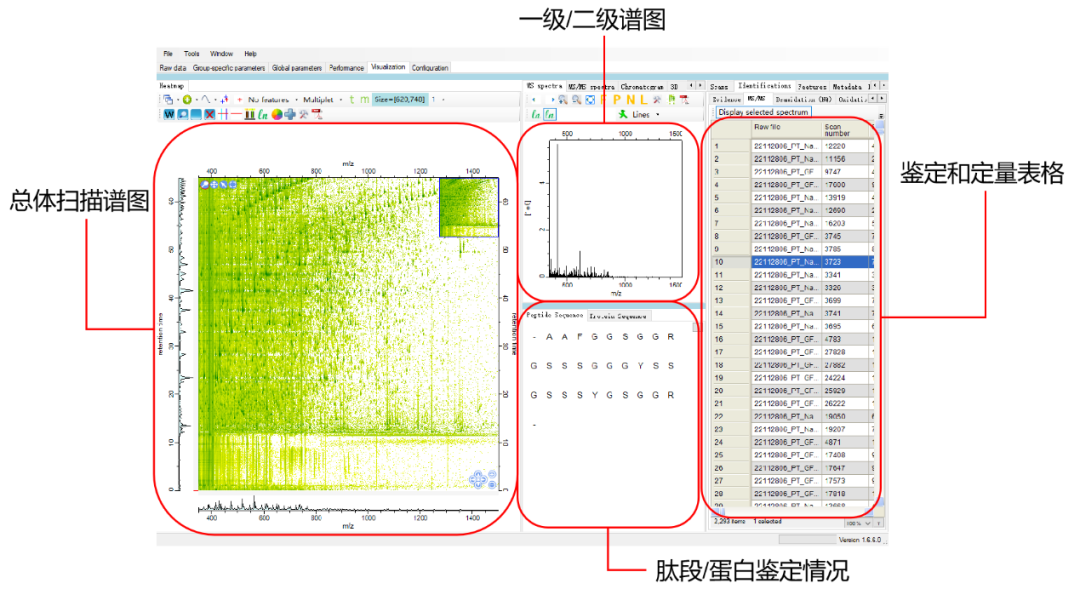
4.结果解析:MaxQuant运行完成后,结果文件将存储在指定的输出文件夹中。主要的结果文件是proteinGroups.txt,它包含了每个蛋白质的量化信息。其他结果文件,如peptides.txt和evidence.txt,包含了更详细的信息,如肽段和PSM(Peptide Spectrum Matches)的信息。

相关技术文章分享:
蛋白质质谱鉴定
蛋白分子量测定
蛋白质结构鉴定
更多科研干货,实验资讯,欢迎关注“百泰派克蛋白质组学多组学”公众号 |
|

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 






 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 发表于 2025-3-9 05:24
发表于 2025-3-9 05:24


 发表于 2025-3-9 05:25
发表于 2025-3-9 05:25