金桔
金币
威望
贡献
回帖0
精华
在线时间 小时
|
登陆有奖并可浏览互动!
您需要 登录 才可以下载或查看,没有账号?立即注册


×
学习笔记+相关补充
参考:https://www.bilibili.com/video/BV1C4411w7jM 一、测序原理
1.1 测序原理
这里主要是高通量测序技术(二代)原理,主要是了解其中的一些基本概念,例如文库等 1、如何识别不同的碱基(ATCG)
(1)光信号:通过碱基带有不同的荧光基团(Illumina、PacBio)
- 问题:不同使用末端终止技术,效率低
- 解决:边合成边测序
- 问题:测序后需要把荧光基团切掉,否则会干预下一次信号捕获
- Illumina采用的技术:边合成边测序;可逆阻断终止技术(解决荧光基团问题);双末端测序(PCR限制了测序的读长问题)
(2)电信号:让碱基基团通过电极,由于化学结构不同所以带的电荷也不同,因此产生不同的电信号(Nanopore)
2、测序流程
建库 —— cluster —— 测序
1.2 测序建库
1、文库(library)
(1)定义:DNA片段的集合,将测序片段打断之后就构成了一个DNA文库
(2)顺序:
- 对DNA样品进行随机打断(机械、超声波、媒介等方式),其中500bp文库是峰值,意思是里面大部分bp片段是在500左右
- 电泳,将在一定范围的DNA进行回收(如果是500bp文库可以回收300-800bp长度的片段),也叫做插入片段长度后面会用
- 文库构建
- 加A碱基,进行末端修复,更方便连接接头
- 加测序引物
- 加index标签,给不同的样品加不同的index,方便后续区分
- 加adaptor接头,包括PCR P5和P7接头分别加在序列的两端,与测序芯片上的接头刚好互补配对

图1.1 文库构建后仅中间部分为序列部分
(3)PRC-free文库:GC比例异常的样品<35% 或 >65%的建议使用这种文库
2、测序芯片(Flowcell)
流动池(Flowcell)是二代测序/NGS(Next-Generation Sequencing)使用的芯片类型,在三代测序中使用的是SMRT 芯片(PacBio)和纳米孔芯片(Oxford Nanopore Technologies) 1.3 Cluster
1、作用:原来的荧光信号太弱,这一步是为了放大荧光信号,将相同的一簇放到一起
2、桥式PCR:
- 将P7、P5接头固定芯片上,因为和文库是配对的,所以进行互补杂交后文库的序列就被种到了芯片上
- 加入DNTP和聚合酶,就会从引物开始沿着模板合成一条新的序列,这条DNA序列是和原来互补的
- 加入碱性溶液和中和溶液会拷贝出一条链,经过多次就会生成一簇
这里不是太清晰,只知道是DNA两端加了接头,然后在芯片上被拷贝复制成簇就行了 1.4 上机测序
1、边合成边测序
通过ATGC可以结合的特性,让他们自己进行结合配对。在结合的过程中,每个碱基加了终止基团,结合后就可以根据荧光进行测序了。
2、可逆阻断终止技术
碱基结合后激发荧光检测信号,然后去除阻断或者终止基团 和 荧光基团。然后进行下一个碱基的结合测序(拍照)
3、reads:测序得到的片段称为reads,测完reads后测index得到样本信息
4、双末端测序
Illumina是先测完一条序列,然后再测互补的那条序列
5、数据处理流程
因为测序得到的不是直接的序列,而是荧光照片,还需要进行图像处理。
(1)Base Calling:存为bcl文件
- 原理:每次测图片中的相同位置的荧光,得到序列信息(最简单的方法,实际上比这复杂,跟波长有关)
- 输出:得到bcl文件,存储坐标、序列、质量等信息
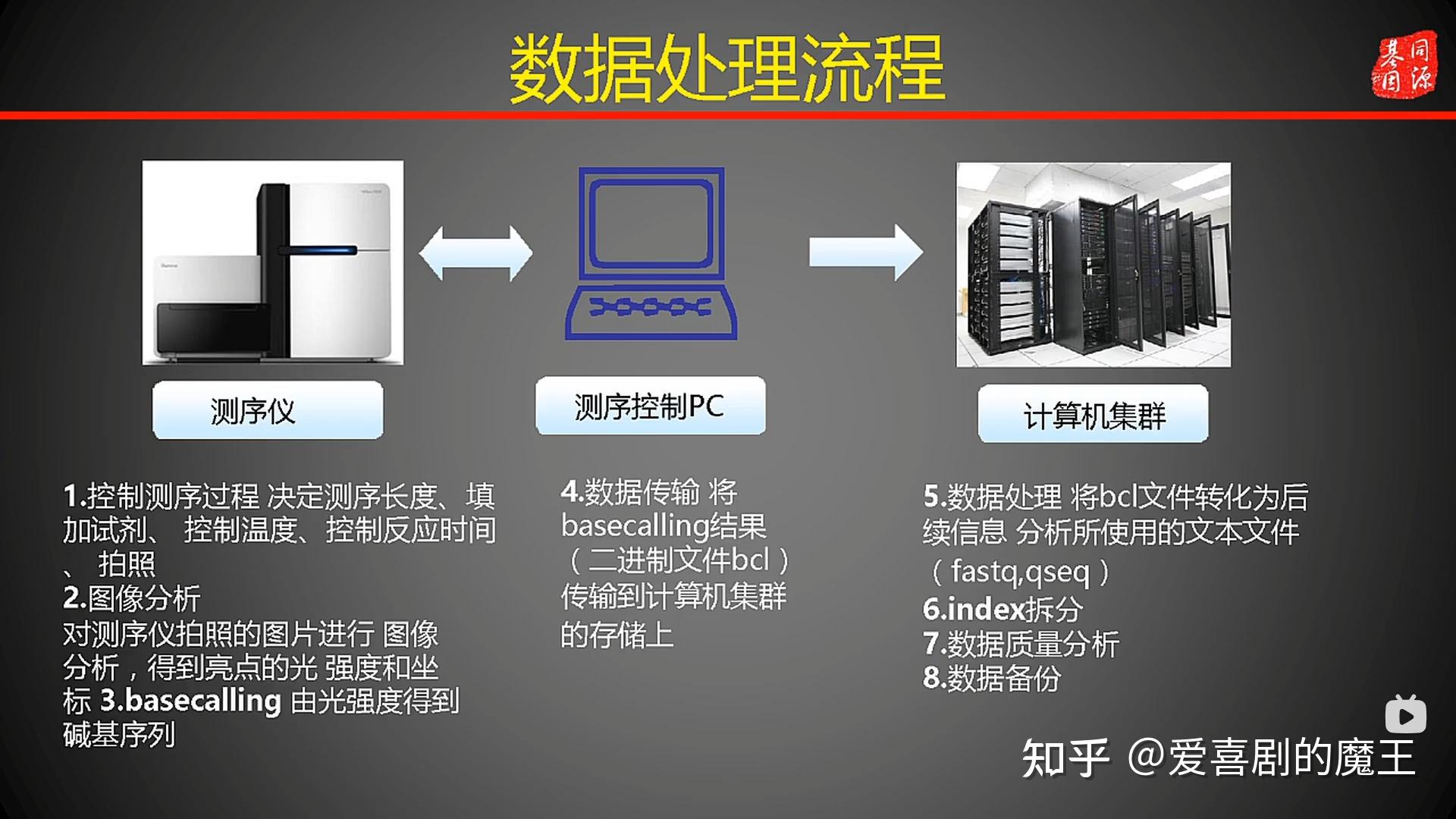
图1.2 数据处理流程
1.5 大片段文库
1、定义:大片段文库指文库长度大于1k的片段,1k一下的称为小片段文库
2、目的:获得reads之间的物理距离关系
3、问题
(1)无法PCR太长片段
(2)只测序很短序列的情况下,合成大片段会造成资源的浪费
4、方式:环化处理,然后进行打断,选取生物素标记的片段,然后再按照原来的小片段的步骤来
5、pairend和matepair
pairend是小片段文库双末端测序,matepair是大片段文库的双末端测序
三代测序(PacBio 和 Nanopore) 本质上不直接使用传统的双端测序技术,但它们通过生成长读长来提供相当于双端测序的上下游信息。 6、文库方向:两种方式的文库方向是不同的,具体没太明白也不太关心
1.6 fastq文件格式
1、文件拓展名:fq或者fastq,相比与fasta多了质量信息
2、格式:分为4行
- 第一行:以‘@’开头,是这一条read的名字,这个字符串是根据测序时的状态信息转换过来的,中间不会有空格,它是每一条read的唯一标识符,同一份FASTQ文件中不会重复出现,甚至不同的FASTQ文件里也不会有重复;
- 第二行:测序read的序列,由A,C,G,T和N这五种字母构成,这也是我们真正关心的DNA序列,N代表的是测序时那些无法被识别出来的碱基;
- 第三行:以‘+’开头,在旧版的FASTQ文件中会直接重复第一行的信息,但现在一般什么也不加(节省存储空间),可能分开看起来没这么混乱吧;
- 第四行:测序read的质量值,这个和第二行的碱基信息一样重要,它描述的是每个测序碱基的可靠程度,用ASCII码表示。ASCII码数值越大,出错率越小,质量值越高。公式为 Q=-10\log_{10}P_{error} , P_{error} 为碱基出错的概率。
- 质量如果为15,那需要加上64转为ascii值为79,用79的ascii值作为质量。因为ascii 33之前都没法用符号表示,64为@所以从这个开始
- 但是好像现在也可以从33开始,做质量控制的时候需要看清楚有没有如!、%、( 等特殊字符,如果有就减去33,如果没有就减去64
@DJB775P1:248:D0MDGACXX:7:1202:12362:49613
TGCTTACTCTGCGTTGATACCACTGCTTAGATCGGAAGAGCACACGTCTGAA
+
JJJJJIIJJJJJJHIHHHGHFFFFFFCEEEEEDBD?DDDDDDBDDDABDDCA
@DJB775P1:248:D0MDGACXX:7:1202:12782:49716
CTCTGCGTTGATACCACTGCTTACTCTGCGTTGATACCACTGCTTAGATCGG
+
IIIIIIIIIIIIIIIHHHHHHFFFFFFEECCCCBCECCCCCCCCCCCCCCCC 3、Q20
(1)T(ASCII=84) - 64 = 20,根据公式Q=-10\log_{10}P_{error},如果Q为20,那么错误率P则为10的-2次幂,P=0.01
(2)代表含义:
- Q20:错误率的百分之一
- Q30:错误率的千分之一
- Q40:错误率的万分之一
1.7 测序饱和度评估
1、测序覆盖率和测序深度
(1)测序深度
测序深度(SequencingDepth),是指测序得到的碱基总量(bp)与基因组大小(Genome)的比值,或者理解为基因组中每个碱基被测序到的平均次数。(将基因组测了几遍, 测序深度能减少假阳性和测序错误率。)
(2)测序覆盖率
测序覆盖比例(Sequencing Coverage)是指测序获得的序列占整个基因组的比例。指的是基因组上至少被检测到1次的区域,占整个基因组的比例。当然,有些文章中也会将测序深度称为Coverage,容易给我们带来混淆。因此,我们还是需要根据语境来推断Coverage的意思。
2、测序覆盖率和深度的关系
(1)碱基平均测序深度为10时,测序覆盖率就能到100%
3、测序不饱和的影响
(1)影响序列拼接
(2)RNAseq定量不准
(3)宏基因组不能准确反映物种组成
1.8 数据质控
1、指标
(1)碱基含量分布
- 测序数据与基因组GC含量一致
- GC含量表示测序数据中ATCG四种碱基的分布,CG含量应该相同,AT含量相同
- ATCG含量的比例与基因组的碱基分布一致
- 测序随机均匀增加,ATGC同时倍增
(2)碱基质量分布
- 首先设置质量值,例如>=Q20为好碱基(Q20是对整体碱基来说)
- Q20百分比:质量值大于等于20的碱基占总碱基的比例
2、FastQC
(1)作用:可以用于评估数据的质量
(2)地址:FastQC A Quality Control tool for High Throughput Sequence Data
在实战中再进行使用和测试,这里先做了解 1.9 测序数据处理
1、去除非基因组本身序列(如图1.1白色之外)
(1)去除adapter
(2)测序引物
(3)barcode
(4)index等
2、数据本身处理
(1)含N碱基过多的reads(N代表未知)
(2)去除低质量数据(一般按照Q20判断)
(3)数据过滤:pairend的reads只要有一个不满足条件就同时去掉两条reads
(4)duplication:两对reads完全一致(是正反测序两条链都要完全一致)
RNAseq和16S测序中duplication不能随便去掉,会造成丰度信息丢失 (5)去除insert size偏差过大的reads(可选)
二、短序列比对
2.1 定义
将测序的reads重新定位到基因组上,也叫做回帖或者mapping
2.2 illumina测序数据特点
(1)测序覆盖全基因组
(2)读长短
(3)测序数据一定错误率
(4)测序数据深度高
(5)测序数据具有pair end关系
2.3 reads比对情况
(1)reads无错配的比对到基因组的唯一位置(perfect match)(1 vs 1无错配)
(2)reads有错配的比对到基因组的唯一位置(1 vs 1有错配)(测序错误或者变异)
(3)reads无错配的比对到基因组的多个位置(1 vs n无错配)(reads太短)
(4)reads有错配的比对到基因组的多个位置(1 vs n有错配)(会删除这种)
2.4 比对的作用
(1)分类

图2.1 短序列比对的作用
(2)reads利用率 = 比对上的reads数 / 总reads数
- 与参考基因组比对,可以衡量样本与参考物种间的同源性
- 数据拼接中评价reads的利用率,越高越可靠
(3)覆盖深度和覆盖比率
2.5 短序列比对软件
(1)算法
- 空位种子片段索引法
- Burrows Wheeler转换法
- Smith-Waterman动态规划算法
(2)过程
(3)软件
- Burrows Wheeler Aligners(BWA):使用BWT算法的aligner工具
- 1、对目标序列建立索引:bwa index
- 2、reads建立索引:bwa aln
- 3、进行比对:bwa sample
- Short Oligonucleotide Analysis Package(SOAP)中的SOAPaligner/soap2
- 1、建立索引:2bwt-builder(也是用的bwt算法)
- 2、进行比对:soap xxx
- bowite:常用于RNAseq分析
- 建立索引:bowtie-build
- 比对:bowtie2 xxx
BWT是一种数据压缩算法,通过轮转的方式压缩序列:BWT算法原理解读-CSDN博客 2.6 sam文件格式
爱喜剧的魔王:生物信息学入门——BAM文件的格式 2.7 估计insert size
(1)定义:插入片段insertsize大小也就是文库片段的长度,同样也是两条测序reads的物理距离
(2)计算insert size:insertsize=reads2_start_pos + reads2_len - reads1_start_pos

图2.2 计算inset size大小
2.8 RNAseq基因差异表达
(1)mRNA加工
- 去掉两侧UTR区域
- 切掉内含子(Intron):是间隔序列,无编码区域
- 不同RNA可能还会发生可变剪切
- 真核需要在3‘端添加Poly A尾(A碱基)
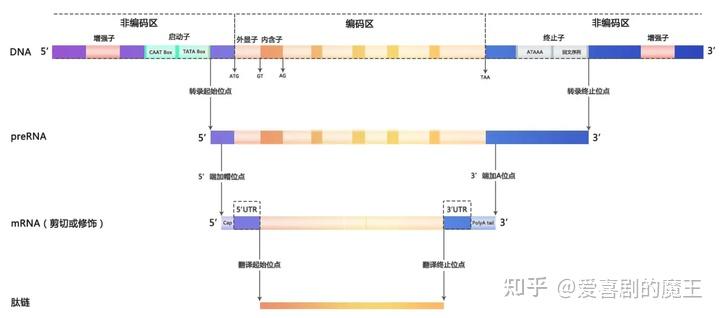
图2.3 DNA到mRNA加工结构示意图
(2)表达量计算
- RPKM:基因表达量计算使用RPKM法(Reads Per Kb per Million reads),
- 公式: RPKM(x)=\frac{C(x)}{\frac{N}{10^{6}}\times\frac{L(x)}{10^{3}}}=\frac{10^{9}\times C(x)}{NL(x)}
- C:Reads Count:在基因A或转录本上获得的reads数目。
- \frac{N}{10^{6}} :Total Reads:在整个样本中获得的reads总数,除以10的6次方是为了将计算单位转为每百万的平均
- \frac{L}{10^{3}} :Transcript Length:基因A外显子区域包含的碱基数(bp),除以10的3次方是为了转为kbp
- 代表含义:用来衡量每百万reads 每一千碱基对 中包含的reads数(被测序的次数),从而估算基因的表达水平。
- 问题:可变剪切reads无法对比回去
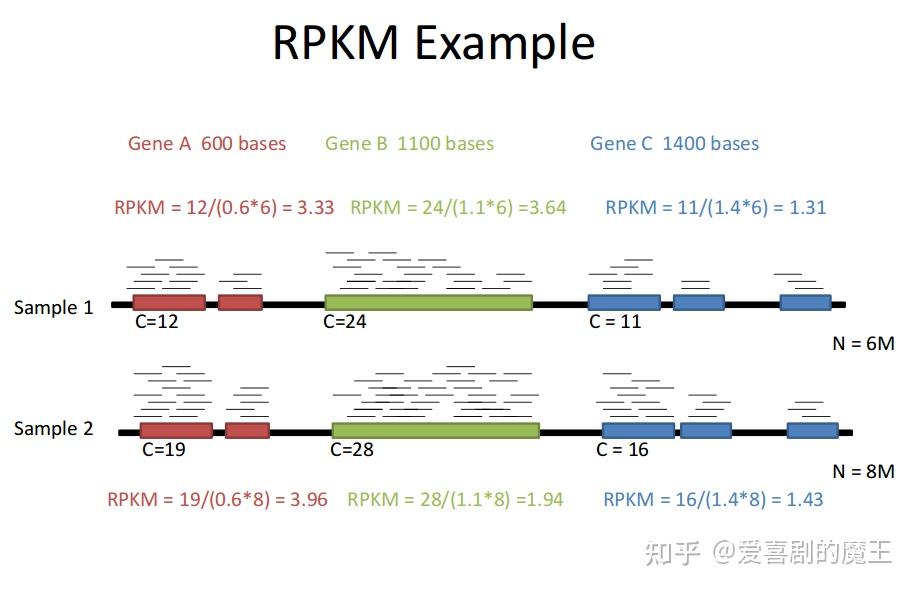
图2.4 RPKM的计算示例
- FPKM(Fragments Per Kilobase Million),每1百万个map上reads中map到外显子的每1k个碱基
- 公式: FPKM=\frac{total\,exon\,Fragments}{mapped\,reads(Millions)\times exon\,length(KB)}
- 计算:与RPKM基本一致,将C换为mapped到外显子的片段数量
- TPM(Transcripts Per Million)
- 意义:在TPM中先消除基因长度的影响,再消除测序深度的影响
- 公式: TPM(x)=\frac{C(x)}{Length(x)}\div\frac{\sum_{i}^{n}{\frac{C(i)}{Length(i)}}}{10^{6}}=\frac{\frac{C(x)}{Length(x)}\times 10^{6}}{\sum_{i}^{n}{\frac{C(i)}{Length(i)}}}=\frac{RPKM(x)}{\sum_{}^{}{RPKM}}
(4)比较基因的差异表达
- FoldChange
- 通过FDR矫正
- 对每个基因进行p-value的计算(公式太复杂了)
- FDR=E(V/R)
这里应该有现成的工具计算,具体的值含义就不看了 2.9 变异检测
1、变异类型
(1)SNP:单碱基
(2)SV:结构变异
2、比对细节
(1)无覆盖
(2)目标序列被覆盖,但只有一种碱基覆盖(√)
(3)目标序列被覆盖,只有一种碱基覆盖,但覆盖碱基与目标序列碱基不同(×,可能是变异位点)
(4)目标序列被覆盖,只有一种碱基覆盖,但覆盖碱基与目标序列碱基不同(多种碱基类型)(×,可能是错配,也有可能是测序错误造成)
- 处理有歧义的位点:
- 过滤掉小于Q20
- 去除落在重复区域的位点
- 去掉低频位点
- 使用概率模型进行过滤
3、检测插入缺失:利用pair end
2.10 物种组成与丰度分析
1、环境微生物
2、16s高变区测序
(1)一共有9个高变区,越长越好,对于物种鉴定有帮助
(2)测序数据不能去除duplication,否则丰度信息会丢失
(3)reads拼接为tags(pair end reads拼接)
(4)物种分类
2.11 FAQ
1、建立索引错误
(1)序列不能太短,否则无法建立索引
(2)序列文件中不能有回车符
(3)选择正确的bwa index选项
2、短序列比对对资源的消耗
3、提高比对效率
(1)完善软件算法
(2)提高硬件资源
(3)比对前进行数据处理
(4)数据拆分合并提高效率
4、matepair文库
(1)soap中要用-R选项
(2)bowtie比对中加--fr或者rf、ff指明read方向
5、短序列比对和长序列比对
(1)长序列是对比多少的问题,短是比对有无的问题
(2)长序列可以容许更多的gap和错配
(3)亲缘关系太远无法使用短序列比对,比对结果不好
三、序列拼接
3.1 序列拼接简介
有的地方也叫de novo拼接(从头拼接)

图3.1 序列拼接的地位
1、序列拼接存在的问题
(1)两条序列的方向
(2)overlap大小
(3)overlap之间存在错配
(4)一条序列与多条序列之间存在overlap
(5)连接之后是否可以继续连接
2、相关名词
(1)reads
(2)pair end和mate pair
(3)insert size
insert size其实就是二代测序中去除接头的部分,当insert size较小时pair end(双端)测序就有可能出现测通的情况,将不属于序列的接头也测进去;当insert size较大的情况下,也有可能出现没有reads重叠的情况(inner mate)。这个时候对于序列拼接可能会造成影响,可以通过大片段文库(insert size)对reads的物理位置进行定位。
参考:
黄树嘉:一篇文章说清楚什么是“插入片段”?
https://guangchuangyu.github.io/cn/2014/08/insertion-size/ (4)kmer
Kmer为一段长度为k的DNA片段,是由测序reads剪切一部分得到的。k为一个奇数,k=几,就为几mer。
可以获得:length - k + 1条k-mer序列
参考:爱喜剧的魔王:生物信息学入门——基因及相关基础知识 (5)contig
拼接软件基于reads之间的overlap区,拼接获得的序列称为Contig(片段重叠群)
准确来说时利用kmers连接成contig
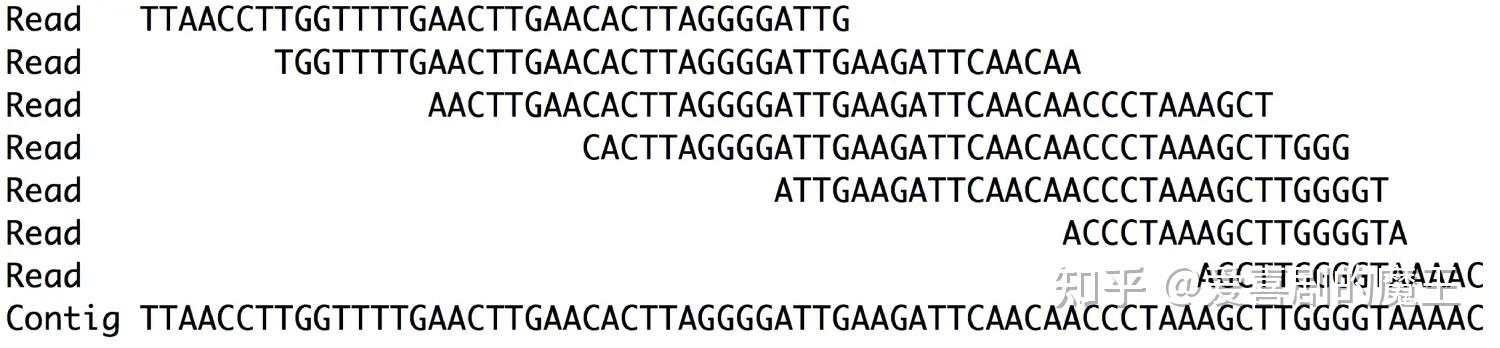
图3.2 contig示意图
(6)scaffold
根据配对信息将contig连接成更长的片段叫做scaffold 3.2 pregraph
1、可用数据
(1)两条pair end关系reads
(2)reads之间具有overlap
(3)reads间具有pairend关系
2、两种算法
(1)overlap-layout-consensus(OLC):适合长reads
(2)德布鲁因图 (De Bruijn graph):适合短reads
3.3 基于De Bruijn graph的短序列拼接步骤
(1)构图
- 将reads切割成k-mer,k一般为奇数
- 将原reads反向互补再用相同方法取kmer
为什么切kmer,直接用reads进行序列拼接,可能会导致错误加到拼接的序列中。
通过kmer计数的方式
去掉kmer频数为1的(因为高通量中这种只测一次情况是不可能的,有可能是测序错误) (2)构建contig
- 构图:两个kmer间只是有k-1的overlap才可以连接起来(用有向图)
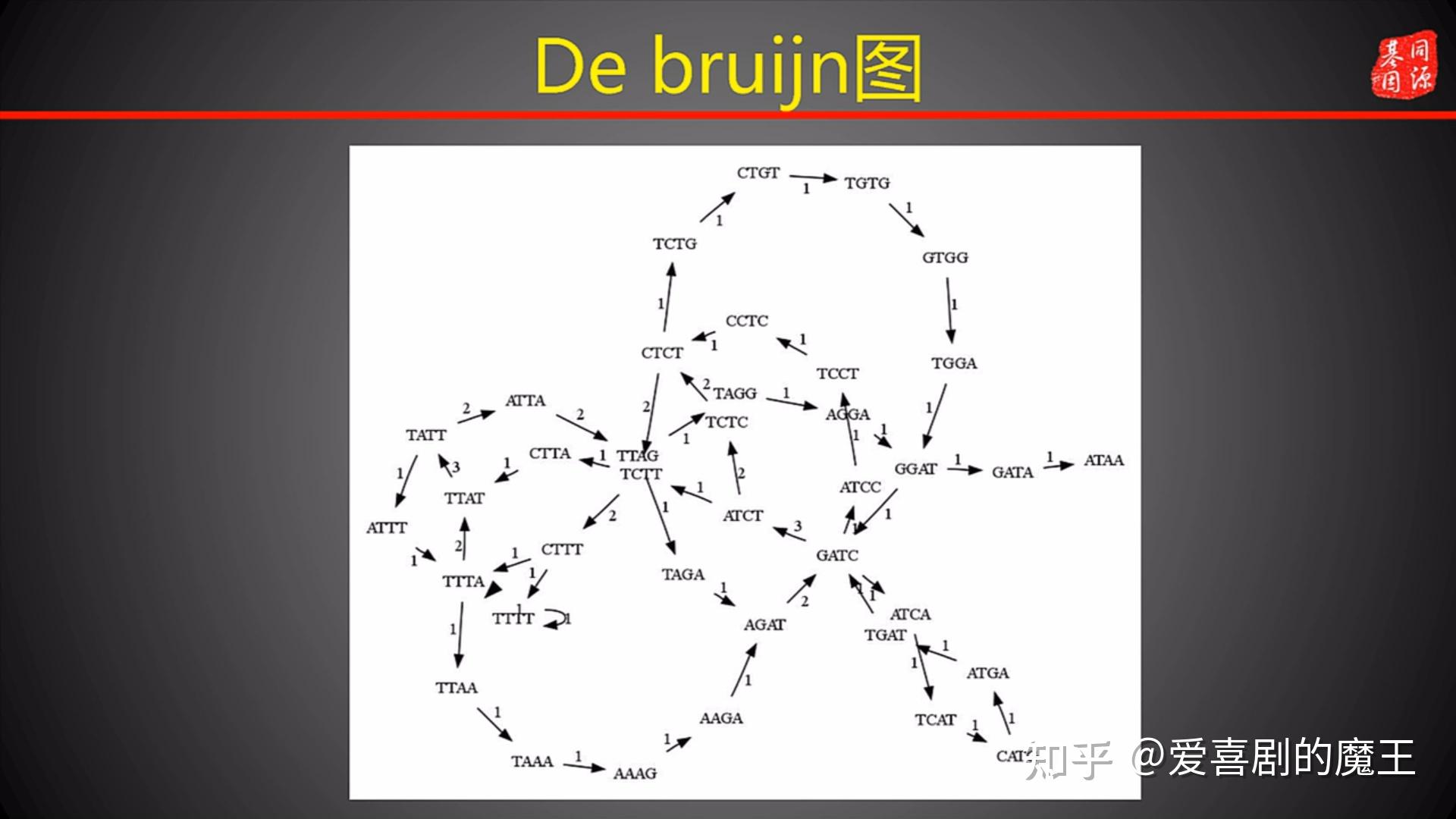
图3.3 构建De bruijn图
- 简化图:
- 合并node:就是将无分叉的node合并到一个里面
- 去除tips:去掉小的分叉
- 合并bubble:随机取一个碱基(bubble是因为有两个碱基不同,其他都相同导致的)
- 解repeat:repeat是重复序列分布在不同位置
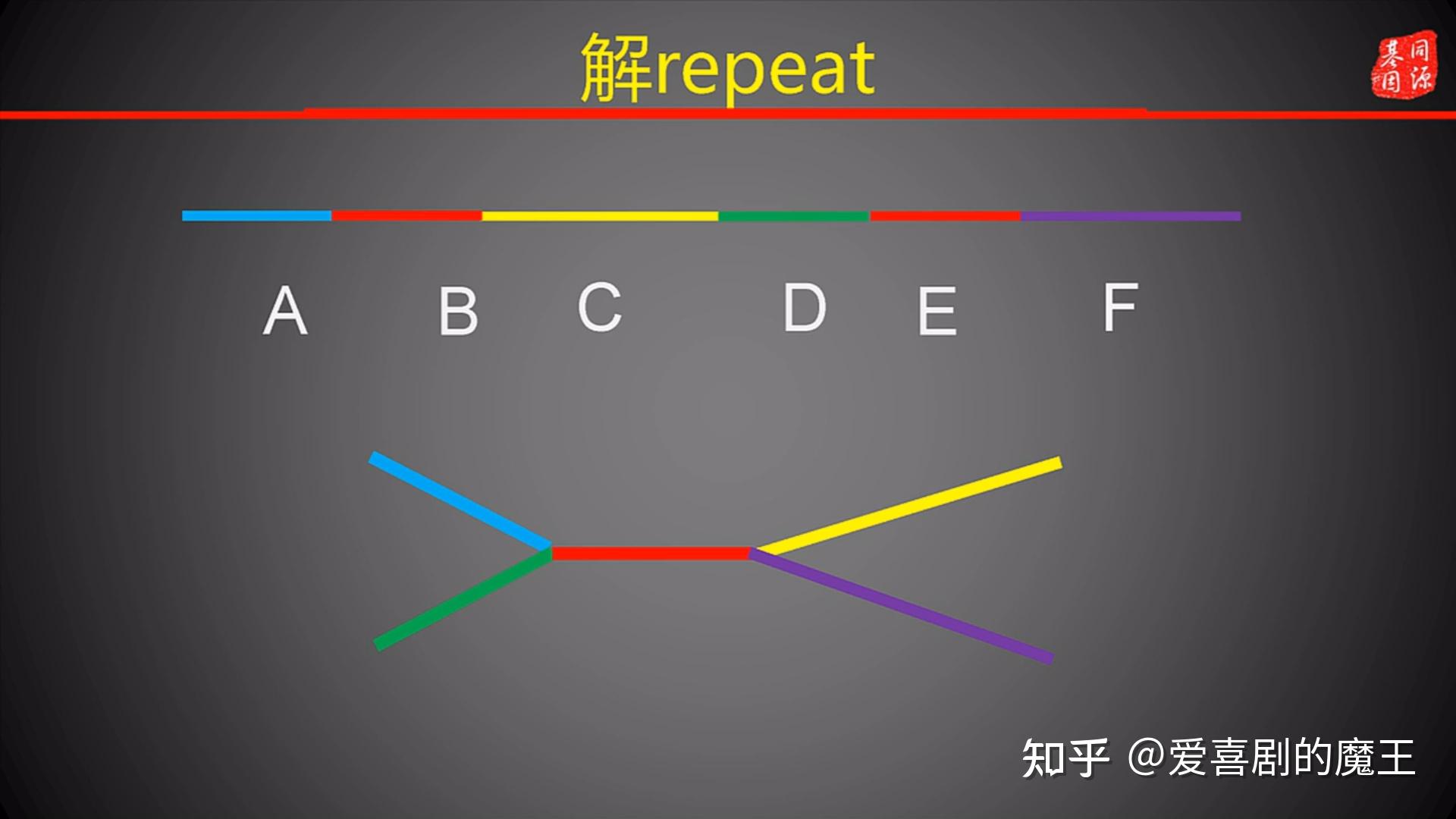
图3.4 重复序列导致的问题
- 输出contig,contig之间没有overlap
常见名词
1. nodes:每个kmer称为一个点/node,理论上nodes数和基因组数大小差不多,过高可能是数据错误率高或者存在杂合,过小kmer也多可能存在重复度
2. edge:点的连接,越多越好
3. tips:点的连接存在分叉,有的分叉无法继续连接下去,称为断头或者tips。理想情况下就俩,头和尾
4. bubble:kmer分成两个路后分别延伸,后面又回到同一条路上变成环状,称为气泡或者bubble。可能是基因组上的杂合位点导致的
------------------
AAAA
↓ ↑
AAAG
------------------ (3)构建scaffold(包括map)
通过reads之间的pair end或者mate pair关系将contig连接成更长的序列,称为scaffold
- 利用mapping定位reads和contig的位置(短序列比对)
- 两条reads落在不同contig上用来构建scaffold,一般至少有3对以上reads成功比对才能确定contig之间的关系
- 对比时的情况
- read1正向比对到contig1,read2反向回补比对到contig2上,说明两条contig的方向一致
- read1和2同方向比对到两条contig上,说明两条contig方向相反是互补的
- 将多个contig组装成scaffold,中间的gap补N
第三代长读可以覆盖这些gap
图3.5 mapping示意图
- 重复序列引起的问题
- 重复序列太长,pairend太短,导致无法确定contig之间的关系
- 一条contig与多条具有关系
- 解决:利用matepair文库
- 拼接策略
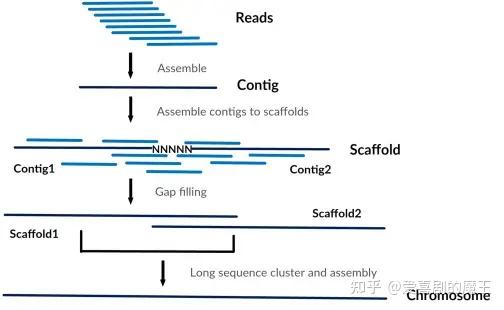
图3.6 基因组序列从头组装示意图(图片来源:Guo et al. Genomics, 2017)
(4)补洞(gap)
使用inset size值(read长度)减去两侧已有区域碱基数,得到gap数目,填补N碱基
- 关于N区域
- N碱基数目不完全准确
- N来自基因组复杂区域,不容易拼接
- 补洞策略
- 利用sanger测序长片段补洞
- 利用洞的上下游设计引物,再使用pcr技术从剩余样本中进行扩增,然后pcr出来的直接填
- 利用pacbio测序长片段补洞
- 利用pairend关系reads补洞
- 常见问题
3.4 序列拼接软件
1、SOAPdenovo
(1)华大开发
(2)作用:适合短序列拼接,尤其是illumina数据
2、velvet
(1)特点:支持fastq、bam等,不善于连接scaffold,不支持metapair文库
3、SPAdes
(1)特点:适合多种数据之间的混合拼接,例如pacbio、nanopore、illumina
4、Newbler
(1)特点:454测序数据拼接,不开源
5、Miseq PE 300
3.5 基因组污染分析
1、污染来源
(1)序列拼接结果中存在污染,说明测序reads中就存在污染
(2)测序reads中存在污染,说明建库测序中存在污染
(3)在样本提取之后就存在污染
2、基因组污染特征
(1)基因组明显偏大
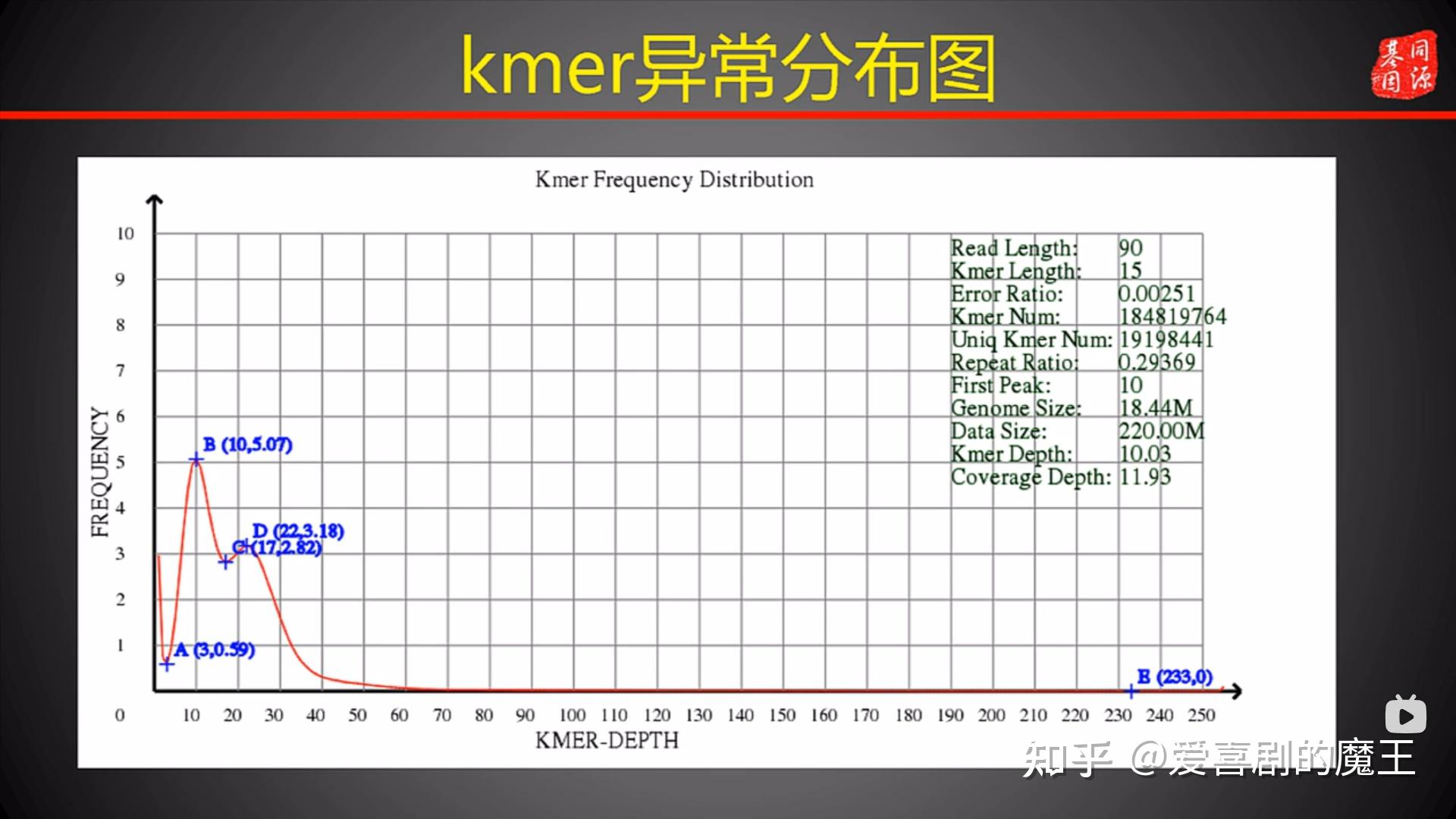
图3.7 kmer异常分布图
(2)序列丰度不同
(3)GC异常
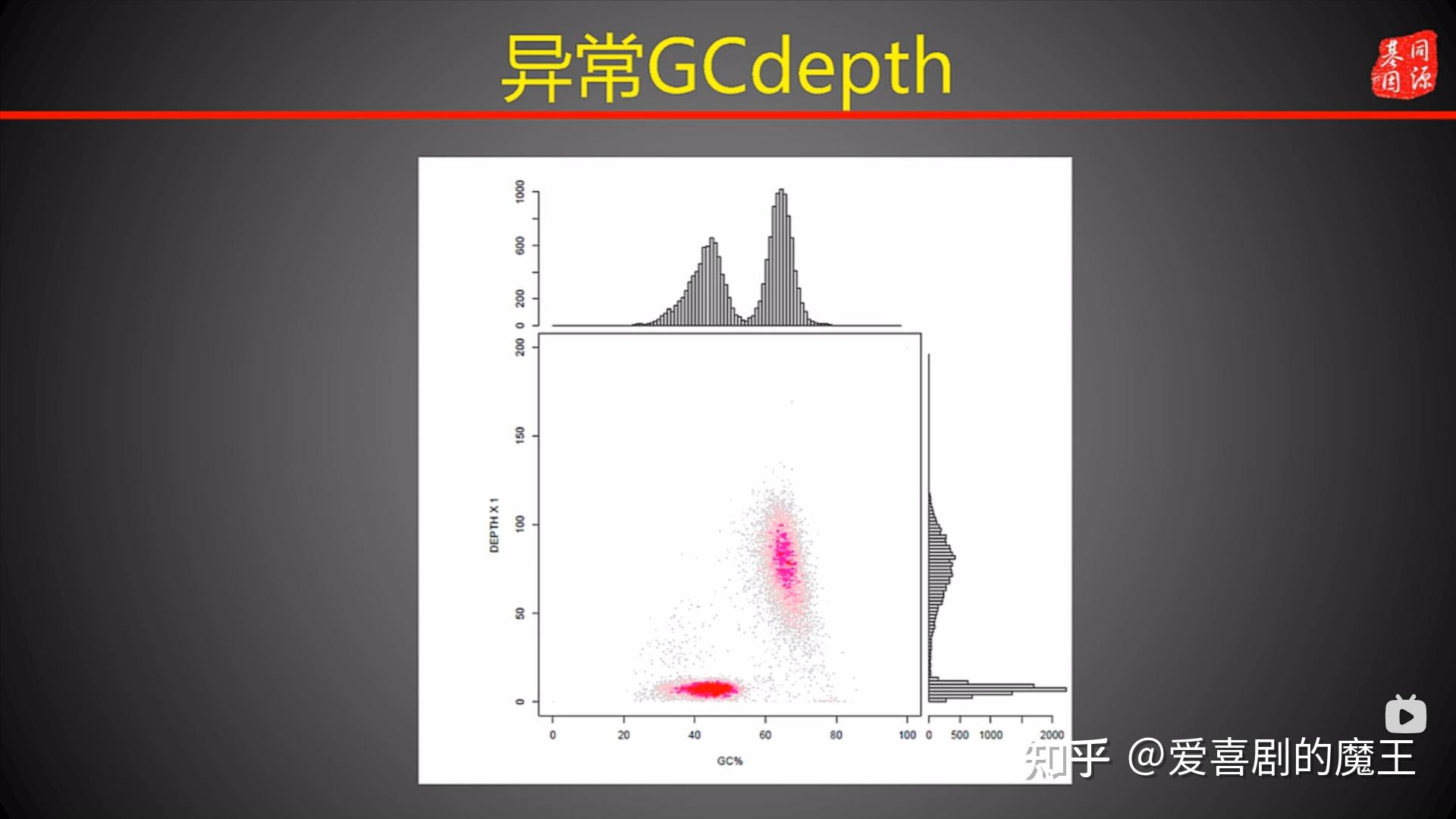
图3.8 异常的GC depth
同时确定是基因组明显偏大 和 GC异常就可以确定基因组污染 3、序列唯一性:序列越长唯一性越高
4、污染鉴定
根据GC分成两个物种,然后把图3.8上面的那部分拿去鉴定 (1)与NCBI进行比对鉴定
(2)预测16s(18s)进行鉴定
5、污染处理:建议重新提取拼接
3.6 RNA seq与meta序列拼接
1、DNA测序与RNAseq比较

图3.9 DNA测序与RNAseq比较
2、RNA seq拼接流程

图3.10 RNA seq拼接流程
拼接出的是转录本的完整序列,因为基因上有一部分是不表达的,mRNA也会加工 3、RNA序列拼接软件
(1)Trinity
(2)oases
(3)SOAPdenovo-trans
4、注意事项
(1)从拼接结果中获取Unigene
(2)拼接要去除duplication
(3)表达定量时不能去除duplication
5、宏基因组拼接
(1)定义:宏基因组(Metagenome),也称为微生物环境基因组Microbial Environmental Genome,或原基因组
(2)问题:拼接前无法准确知道大小和物种情况
(3)解决
(4)序列拼接软件
3.7 FAQ
1、影响拼接的因素
(1)内因:多倍体,基因组杂合,高度重复,低复杂度,GC偏差等
(2)外因:测序数据量,测序质量,文库大小,kmer大小,基因组自身,拼接软件,阈值设定
2、改善拼接结果

图3.11 改善拼接结果
3、如何选择kmer大小
- reads准确度越高,选择大kmer较好
- reads错误率高,选择小kmer,reads利用率高
- 基因组本身特点
4、kmer为什么只是奇数
四、基因组分析

图4.1 基因组分析
4.1 基因预测
1、定义:基因预测是指通过计算方法和算法,从基因组序列中识别和预测可能的基因位置及其功能的过程。
2、基因预测方法
(1)利用软件对物种基因组之间进行预测
(2)通过同源序列比对,和已知近源物种基因集进行比对,将同源比对区筛选处理,作为基因

图4.2 方法比较
3、开放阅读框
开放阅读框(ORF)是一个能够被翻译成蛋白质的核苷酸序列,它从起始密码子开始,直到终止密码子为止。
CDS:coding sequences (编码区)
它就是与蛋白序列一一对应的DNA序列,并且序列中间不存在其他与蛋白无关的序列,和真实情况最接近
举个例子:
例如这个序列:AACGCATGCAGC
如果用预测的方法得到ORF,它会先找到ATG;然后会以中间的字母T为核心,推测三种可能:
第一种:T就是在中心,即ATG,然后按每三个一组,得到:CGC、ATG、CAG
第二种:T在左侧,即TGC,同样得到AAC、GCA、TGC、AGC【这就是真实的CDS组合】
第三种::T在右侧,即CAT,得到ACG、CAT、GCA
还因为DNA双链,所以总共有32=6种不同密码子组合方式*

图4.3 开放阅读框
4、原核生物的基因结构
(1)原核生物基因结构是从5‘端启动子区域开始,到3‘端终止区域结束。
(2)基因转录开始由转录其实位点开始,如何直至终止位点结束,内容包括5’端非翻译区、开放阅读框和3‘端非翻译区
(3)基因翻译是从起始密码子到终止密码子,翻译对象为两个密码子间的开放阅读框区域。
5、常用软件
- glimmer3:使用隐马尔可夫模型’
- prodigal
6、密码子表
要根据具体物种选择不同的密码子表
7、真核生物的开放阅读框
(1)包括外显子(exon)和内含子(intron),内含子将开放阅读框分为若干个小片段
(2)外显子和内含子之间存在GT-AG规律
- 内含子序列5‘端的起始两个核苷酸总是GT,且3’端最后两个核苷酸总是AG,即:5‘-GT……AG-3’
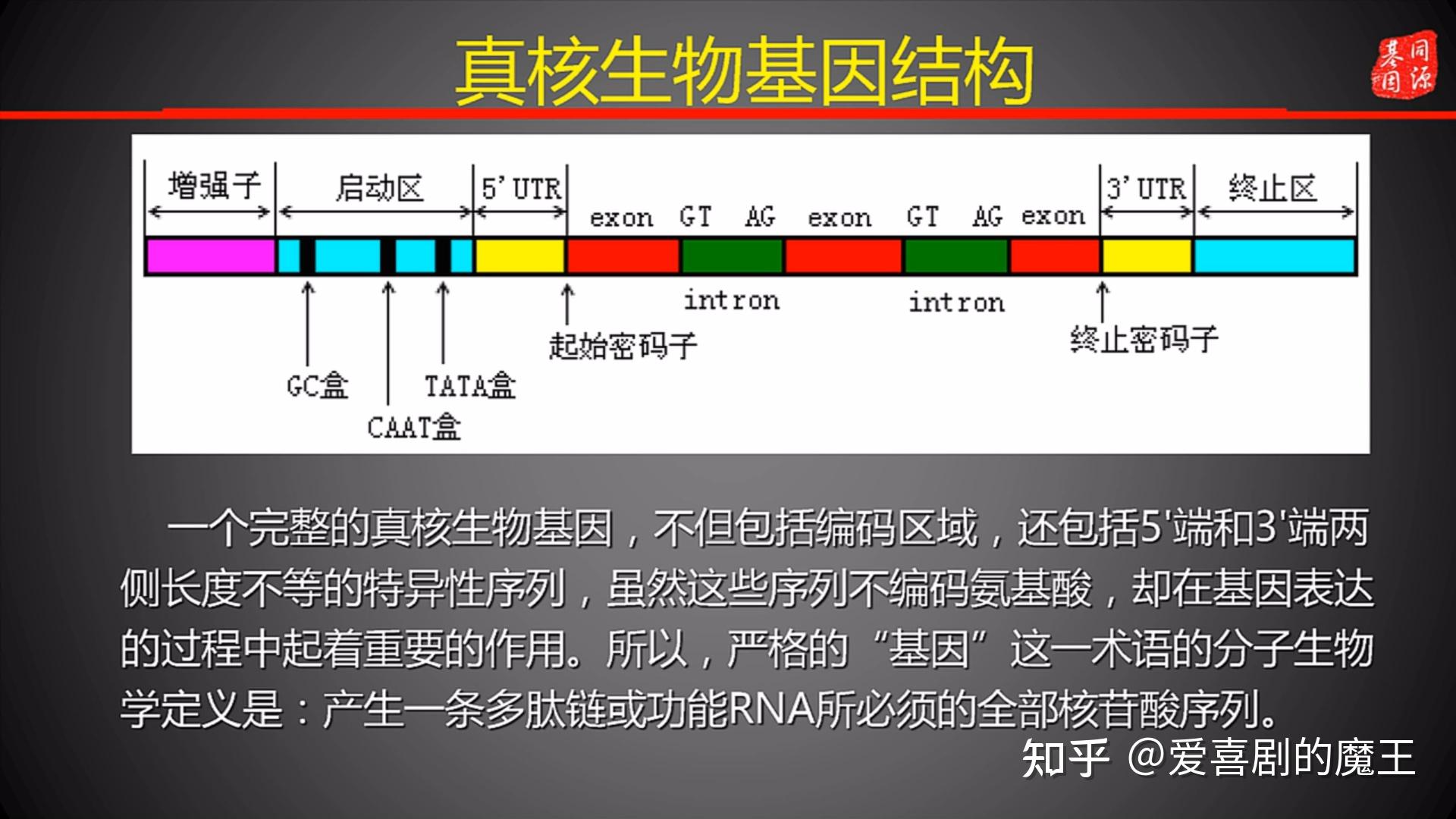
图4.4 真核生物基因结构
真核生物可以用Augustus进行基因预测,或者从头预测工具 8、宏基因组基因预测
4.2 基因功能注释
Encode计划,弄清楚每个基因的功能 1、基因功能注释操作:使用基因与数据库进行比对
2、基因功能数据库
(1)基因序列fasta
(2)基因功能信息
3、blast比对阈值设定
BLAST(Basic Local Alignment Search Tool)是一种用于生物信息学的常用工具,用于在数据库中搜索序列相似性的算法。 (1)U被替换成X:氨基酸比对中不存在U,所以blast把他换为X
4、基因功能分类
COG数据库,是Clusters of Orthologous Groups of proteins的简称。,蛋白相邻类的聚簇。
该数据库是对细菌、藻类和真核生物的21个完整基因组的编码蛋白,根据系统进化关系分类构建而成的。对于预测单个蛋白质的功能和整个新基因组中的蛋白质的功能非常有用。
其他还有:
Gene Ontology(GO)数据库
Kyoto Encyclopedia of Genes and Genomes(KEGG)数据库 将样本的基因与COG数据库进行比对,属于哪个分类
4.3 非编码RNA分析
4.4 小RNA分析
4.5 重复序列分析
1、分类:
(1)散在重复
- LTR,长末端重复转座子(long terminal repeat)
- LINE,长散在重复序列(long interspersed nuclear elements)
- SINE,短散在重复序列(short interspersed nuclear elements)
- DNA转座子
(2)串联重复
- 卫星(satellites)DNA
- 小卫星(minisatellites)DNA
- 微卫星(microsatellites)DNA:短的串联重复,也叫STR
串联重复在变异检测中称为CAV的检测即拷贝数变异

图4.5 重复序列分类
2、重复序列根据长度分类:高度、中度和低度
3、重复序列分析
(1)序列比对
- 一般用RepeatMasker软件
- 串联重复一般用TRF软件(Tandem Repeats Finder)
(2)从头预测
4、重复序列影响:测序或者组装
4.4 基因组特殊元件分析
1、CRISPR
Clustered Regularly Interspaced Short Palindromic Repeats RNA是最近几年才发现的原核生物中的调控RNA。
在线预测:http://crispr.u-psud.fr/Server/ 2、CpG岛分析
(1)CpG岛是指DNA序列上的一个区域,此区域含有大量相联的胞嘧啶C、鸟嘌呤G,以及使两者相连的磷酸酯键(p)
(2)许多 CpG 岛位于基因的启动子区域,这些区域对基因的转录调控具有重要作用
(3)作用
- 基因调控
- DNA 甲基化
- 在 CpG 岛区域,DNA 甲基化(指 DNA 上的胞嘧啶碱基通过添加甲基基团而发生的修饰)可以调节基因的表达。通常,启动子区域的 CpG 岛甲基化水平较低,允许基因转录,而高甲基化状态可能导致基因沉默。
- 基因组印记和X染色体失活
(4)预测工具:CpGplot
http://www.ebi.ac.uk/Tools/emboss/cpgplot/index.htm
http://www.ebi.ac.uk/Tools/segstats/emboss_cpaplot/
3、操纵子分析

图4.6 操作子分析
OperonDB
http://csbl.bmb.uga.edu/DOOR/index.php
http://operondb.cbcb.umd.edu/cgi-bin/operondb/operons.cgi
ftp://ftp.cbcb.umd.edu/pub/data/operondb/ 4、基因岛分析
5、启动子分析
(1)定义:启动子是基因的一个组成部分,是位于结构基因5“端上游区的DNA序列控制基因表达的起始时间和表达的程度。启动子本身并不控制基因活动而是通过与称为转录因子的蛋白质结合而控制基因活动的。
(2)可以使用PromoterScan来预测分析启动子区域,其网址为 :http://www-bimas.cit.nih.gov/molbio/proscan/
4.5 共线性分析
1、定义:共线性分析是用于检测和处理自变量(预测变量)之间的多重共线性问题。在基因组中用于研究两个或者多个基因组是否有较好的同源性。同源性好共线性图会呈现斜率为1的直线,如图4.7。
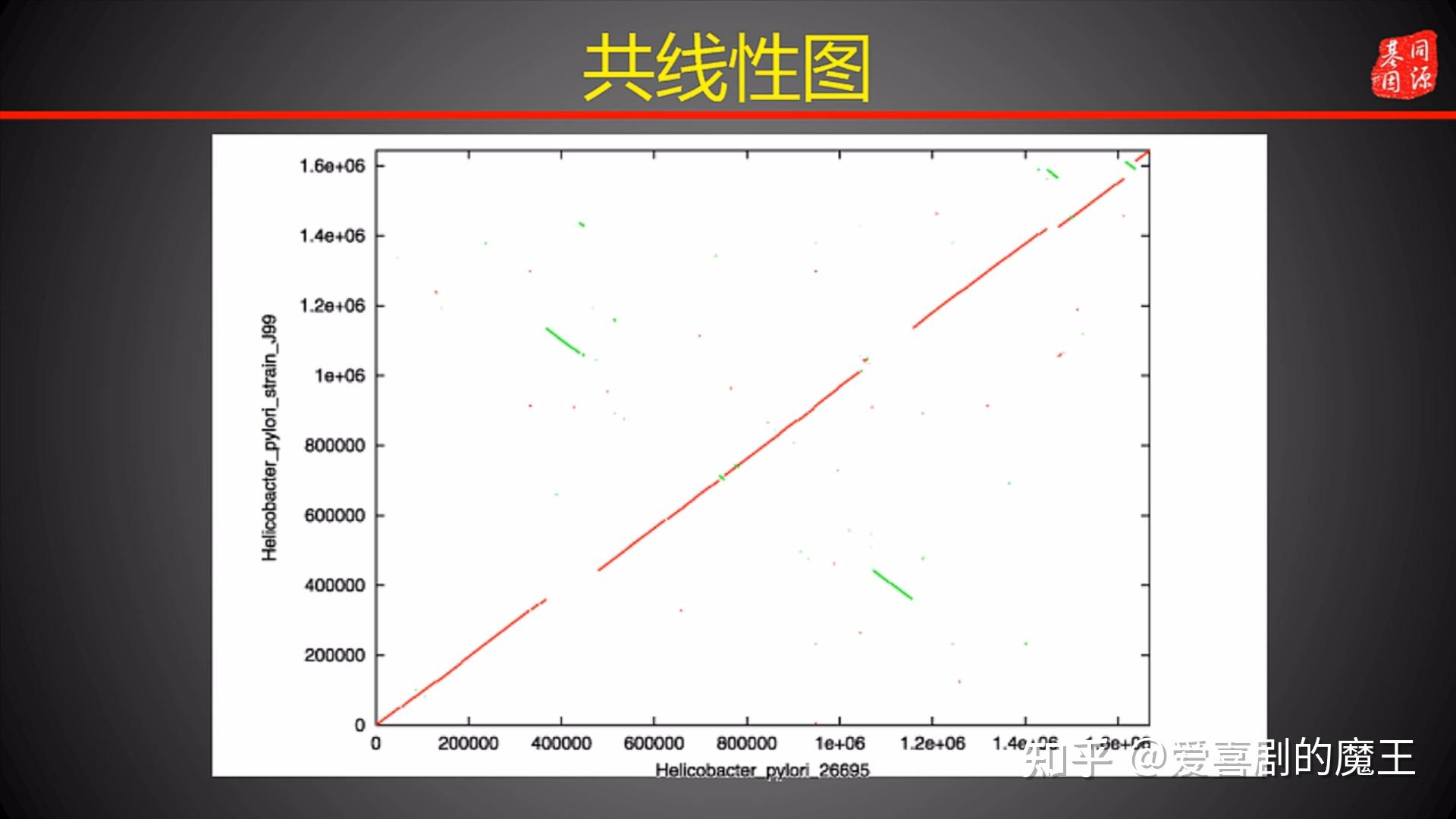
图4.7 共线性图
绿色的可能是互补,斜率为-1
mummer + mummerplot可以生成

图4.8 通过BRIG展示多个基因组的同源关系
BRIG适合同源性小的 4.6 在线序列分析工具
视频展示太快了,有需要后续找吧 4.7 序列比对
1、同源和相似
(1)同源(homology):是指来自于同一个祖先,两个物种从同一个祖先分化后,与不同的环境发生相互作用,其相应的DNA序列将各自发生一些替换或者插入缺失突变,也就是说序列不在精确相同。
(2)相似性(similarity): 是指字符串的相似,并不具有生物学意义
同源一定相似,相似不一定同源
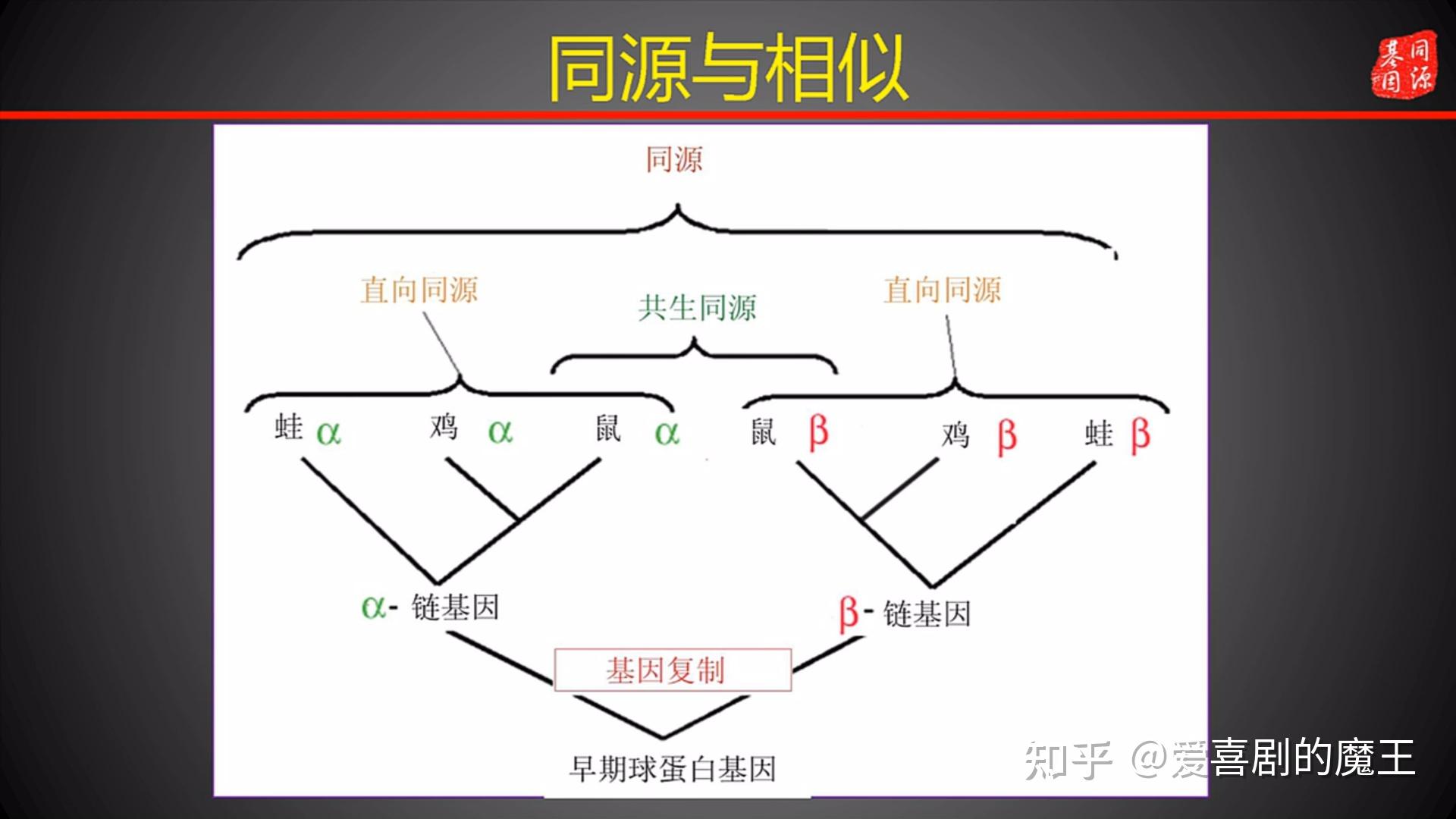
图4.9 同源和相似
2、序列对比需要考虑的问题
(1)比对的对象是什么,DNA还是氨基酸序列
(2)如何判断相似性
(3)哪种算法找到全局或者局部最优匹配
(4)如何判断序列同源性的高低
3、序列比对的情况
(1)碱基类型一致
(2)碱基发生了变化,包括替换、插入删除
4、罚分矩阵
有各种矩阵,需要根据情况选择 5、比对算法
(1)Needleman算法
(2)Smith-Waterman算法
(3)启发式联配算法等。
6、如何评价匹配的显著性
(1)E-value :E值综合考虑了真实匹配与随机匹配的相对概率,序列的长度数据库大小和序列组分的偏向性等数据而计算出来的
4.8 局部比对和全局比对
1、定义
(1)全局比对是用来衡量两条序列整体的相似性,满足整体相似性最大化。
(2)局部比对则不同,两条亲缘关系较远的DNA或氨基酸可能只在一些片段上相似,这就需要找到这些相似性的片段,和其相应的匹配方式。

图4.10 全局和局部示意图
2、局部匹配软件
(1)Blast:Blast全称Basic Local Alignment Search Tool,即"基于局部比对算法的搜索工具"
推出了blast+ (2)FastA:FastA程序采用渐进(heuristic approach)算法将位于同一对角线上相互接近的短片段连接起来。也就是说,通过不匹配的残基将这些匹配残基片段连接起来,以便得到较长的相似性片段。
比上个精确,但是慢 (3)Blat:Blat的全称是The BLAST-Like Alignment Tool,可以称为“类BLAST比对工具“
适合将基因CDS重新定位到染色体上 3、全局比对工具
(1)MUMmer是TRIG在1999年开发的,当时人全基因组还没有发表,只有少部分基因组被测序出来,通过mummer全局比对,第一次看到了不同物种基因组之间的差异。
速度快,资源消耗少,后缀树方式 (2)LASTZ的全称是 Local Alignment Search Tool, blastZ-like,中文翻译就是“类blastz的局部比对搜索工具“
在blastz上的升级 4.9 多序列比对
1、定义:多序列比对(Multiple sequence alignment;MSA),是对三个以上的生物学序列,如蛋白质序列、DNA序列或RNA序列所作的序列比对。
2、构建模型方法
(1)手工比对
(2)同步法:通常对少量较短序列
(3)渐进法
3、多序列比对的特点
(1)多序列比对一般都是将所有的序列放在同一个文件中
(2)每一个样品都会对整体造成很大的影响
(3)序列之间要具有同源关系,并且具有同一方向
4、常用软件
(1)MUSCLE (stands for MUltiple Sequence Comparison by Log- Expectation)
(2)Clutal w
(3)mafft
(4)mauve:输入全基因组文件进行检查
(5)mega
五、变异检测
5.1 变异检测概述
1、遗传和变异
(1)遗传就是子代在这个连续系统中重复亲代的特性和特征的现象。
(2)基因组世代延绵的长期发展过程中,随着时间和空间的变化。结构改变了的基因使生物体发育不同于改变前的性状,于是出现了变异
2、变异类型
(1)单碱基
(2)染色体
- SV
- Corepangene:如果插入缺失发生在基因水平就是基因的获取与缺失
- CNV:片段拷贝数的变异,发生在串联重复区域的插入缺失
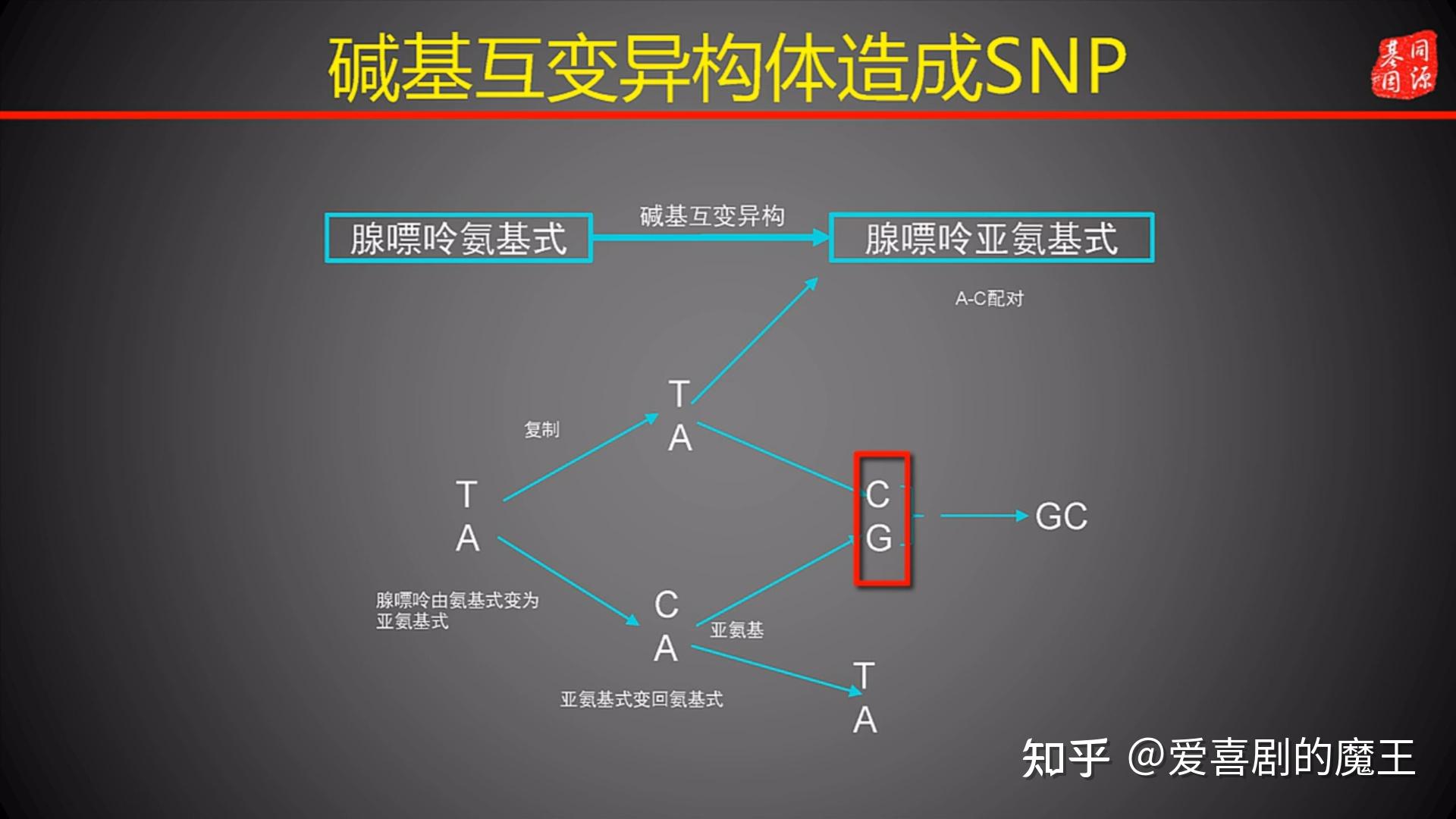
图5.1 碱基互变异构体导致SNP
3、变异出现的主要原因——自发突变
(1)碱基互变异构体导致SNP
- 如图5.1 腺嘌呤(A)正常会和胸腺嘧啶(T)配对,但是变成腺嘌呤亚氨基(A')会和胞嘧啶(C)配对,导致下一次复制时AT变成了CG
- 同样,胸腺嘧啶(T)也可以将AT转为GT
(2)脱嘌呤与脱嘌呤形成InDel
- 碱基偶尔会将核苷酸移除,而留下一个缺口称为脱嘌呤/脱嘧啶,该缺口下一轮复制时不能进行正常碱基配对
- 原因:胞嘧啶(C)的自然脱氨基形成了尿嘧啶(U),导致DNA不能识别所以被除去,形成一个脱嘧啶位点
(3)转座子造成结构变化
转座子(transposon)是一类存在于基因组中的DNA序列片段,具有自我迁移和插入到基因组中其他位置的能力。它们被认为是基因组内的“跳跃基因”。转座子可以插入到基因组的不同位置,可以是基因、非编码区域或其他转座子
- 基因组上的转座子能够随机插入到基因组上
- 如果基因组上存在2个或者多个拷贝,就会发生同源重组,进而导致缺失、重复和倒位等变异
4、变异检测的意义
1. De novo测序:如果一个基因组第一次被测序出来,我们一般称为Denovo测序,主要是需要拼接其基因组;
2. 重测序:已有基因组被发表出来之后,重新测序新的数据,可以不需要拼接,而直接用测序的reads进行短序列比对分析,称为重测序分析。在文献中常常会看到Re-sequencing这个词。其实重测序本质就是找变异。 (1)引起基因组的变化,进而导致基因型或者调控区的变化
(2)根据基因型的变化推测与表型变化之间的关系
5.2 SNP检测
1、定义:SNP(single nucletide polymorphsysm),即单核苷酸的多态性是指等位基因发生了突变,致使不同等位型的出现。
2、Hapmap:Hapmap为国际人类基因组单体型图计划(TheInternationalHapMap Project)是继国际人类基因组计划HGP,之后人类基因组研究领域的又一个重大研究计划。HapMap计划是一个多国参与的合作项目,旨在确定和编目人类遗传的相似性和差异性
3、SNP分类(二倍体)
(1)纯合SNP:纯合SNP是指两个等位基因都发生了同样的突变
(2)杂合SNP:杂合SNP是指两个等位基因中发生了不同的突变
转换是同类碱基的置换(AT→GC及GC→AT),颠换是不同类碱基的置换(AT→TA或CG,GC→CG或TA)。
转换:嘌呤和嘌呤之间的替换,或嘧啶和嘧啶之间的替换。
颠换:嘌呤和嘧啶之间的替换。
SNP在CG序列中出现较为频繁,且多为C转为T,因为C甲基化脱氨基后形成T
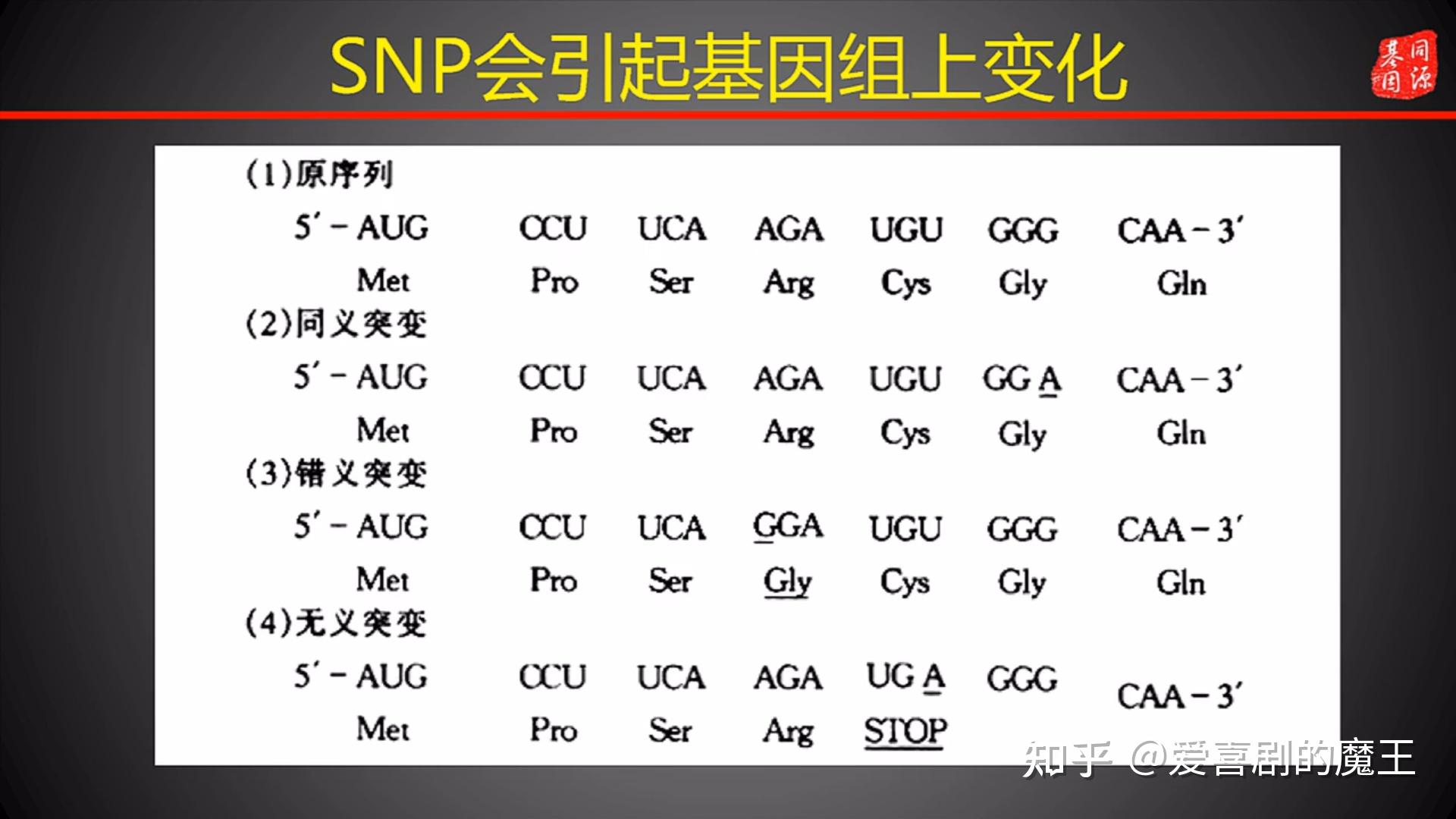
图5.2 SNP导致基因组的变化
同义突变:突变了但没影响氨基酸匹配
错义突变:突变导致氨基酸改变了——镰刀型细胞贫血症
无义突变:突变导致变成了终止密码子,后边的不表达了 3、SNP检测
先保证同源,再检测 (1)利用测序数据与参考序列进行短序列比对
- 优点:
- 1、无需拼接,节约成本;
- 2、包含二倍体信息,可以处理杂合情况;
- 3、利用丰度信息,处理测序错误;
- 缺点:
- 1、reads过短,唯一性差;
- 2、短序列比对只容许很少的错配和空位
- 3、无法解决重复区域问题
(2)利用序列拼接经过与参考序列进行比对
- 优点:
- 1、拼接结果中包含基因组更多的信息
- 2、长序列具有更好的特异性;
- 3、容更多的错配和空位;
- 缺点
- 1、对于二倍体,拼接结果只给出一种情况
- 2、序列拼接存在错误,造成假阳性;
5.3 SV检测
1、定义:基因组结构变异SV是Structural Variation的简称,广义上来说,基因组上所有的变异都可以称为SV。而更狭义的概念,SV特指基因组大片段的插入与缺失。
2、SV检测的问题
(1)基因组本身复杂性;
(2)测序reads读长过短;
(3)insertsize值不确定;
(4)序列拼接不完整,存在N区域等问题等;
3、SV检测方法
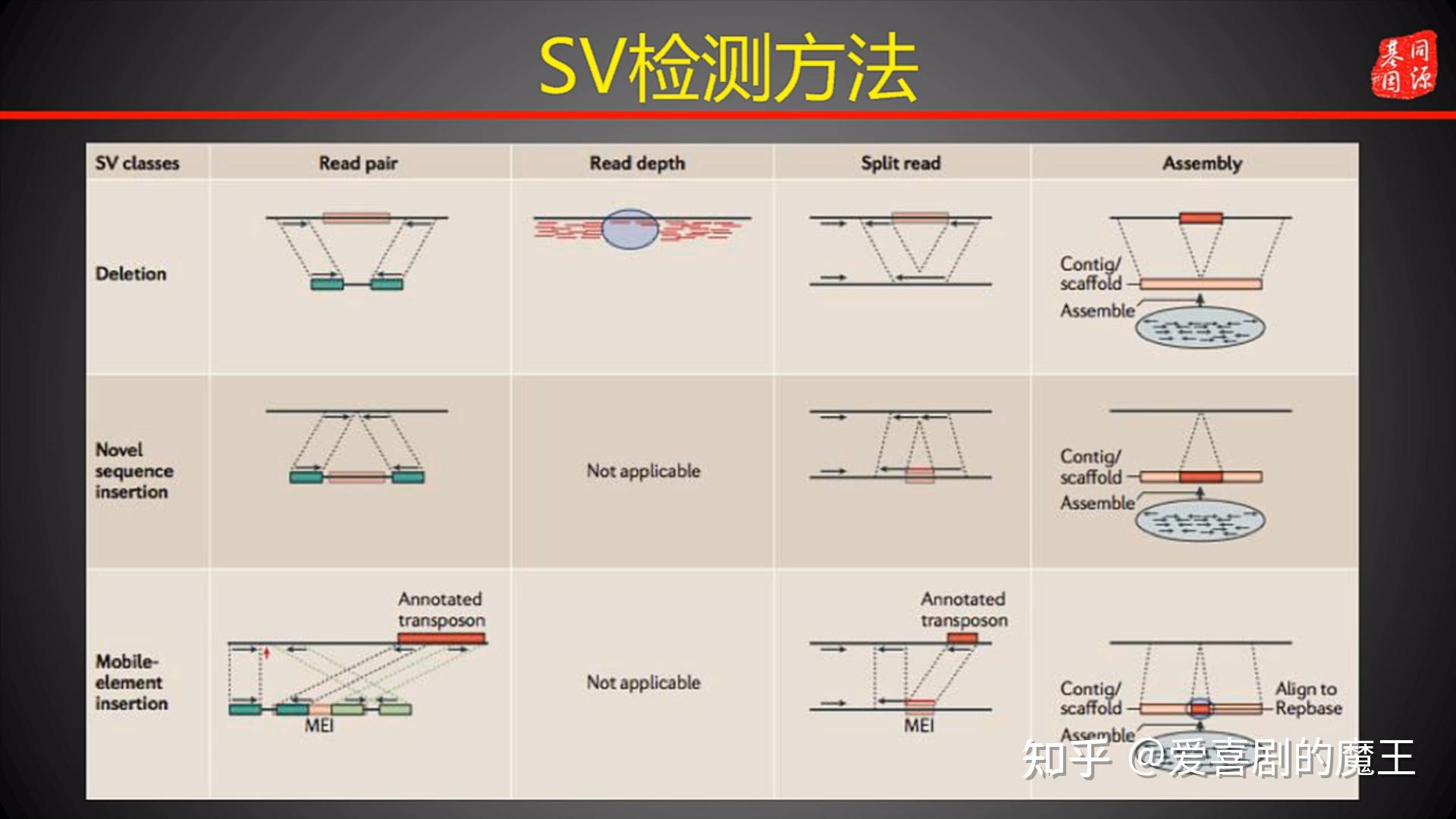
5.3 SV检测方法
(1)基于Pair-end Mapping(PEM):序列插入删除、重复
(2)Split Read(切割read,简称SR)
(3)Read Depth
(4)基于序列拼接的方法
总结
1、对于小的结构变化InDel,或者较长的片段缺失,目前都能够较好地检测出来;
2、对于大多数的插入片段和更复杂的结构性变异情况,例如拷贝数的变化,当前的检测软件基本都没法还解决
3、同时使用read mapping与序列拼接的方法,将结果进行整合,来获得一个相对比较准确的SV检测结果 5.4 CorePanGene集构建
1、定义:CorePanGene(核心泛基因)集是指在不同物种中,经过进化保存的那些核心基因的集合。
Pan-genome: 细菌物种的全球基因库:
Core-genome: 同一细菌物种的所有菌株共享的基因库
Dispensable genome: 存在于同一细菌物种的某些菌株而非所有菌株中的基因库
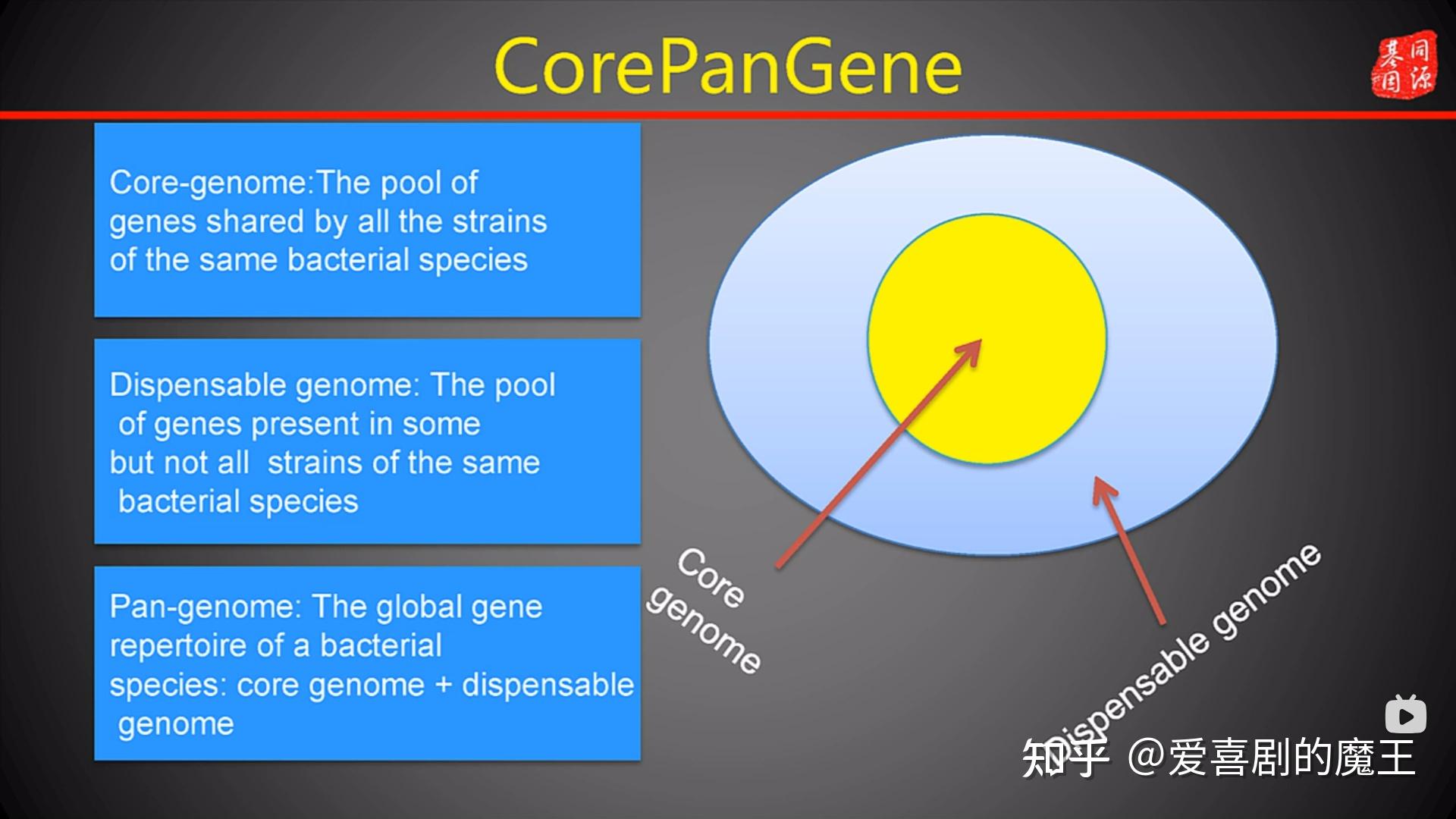
图5.4 corePan gene示意图
2、Dispensable
(1)描述群体特征,亲缘关系等;
(2)筛选特异基因;
(3)基因型与表型关联分析;

图5.4 corePanGene 构建举例
3、常见问题
(1)如何判断两个基因为同源基因,参数设定,阈值选择等
(2)受单个样品影响
(3)组装的不完整性,基因预测的影响
(4)基因组本身的复杂性,多拷贝及基因融合
5.5 变异检测软件
1、短序列比对
(1)soap方法:soap短序列比对——soapsnp进行snp检测——soapindel进行indel检测——soapsv进行sv检测
(2)BWA:bwa短序列比对——samtools进行处理(sam转bam、sort、mpileup)——bcftools进行变异检测
samtools mpileup -ugf H37Rv.fna all.sort.bam -Q 13 -q 0 -F 0.002bcftools view -bvcg -t 0.001 -i-1 -p 0.5 -X 0.01 -b -> al.bcfbcftools view all.bcf |perl vcfutils.pl varFilter -Q 10 -d 2 -D 100 -a2 -w 5 -W 5 -1 0.0001>all.vcf (3)BWA+gatk
(4)indel检测
(3)其他软件
- break
- dancer
- cnvnator
- SSAHA2
- maq
2、基于序列拼接结果SV预测的软件
(1)mummer
MUMmer3.23/nucmer--mum--maxgap=500--mincluster=100--prefix=nucmer ref.fna query.fnaMUMmer3.23/delta-filter -1 -q -r nucmerdelta > nucmer.filterMUMmer3.23/show-snps -C -H -l -T -r -l nucmer.filter>nucmersnp
MUMmer3.23/promer --mum--maxgap=500--mincluster=100 --prefix=promer reffna query.fna
dnadiff reffna query.fna (2)lastz
3、结果处理和验证
1、过虑掉落在重复区域的变异;
2、去除reads支持数为1的低频突变;
3、支持SNP的reads质量值大于Q20;
4、SNP位点附近3bp内没有另外的SNP
5、SNP的覆盖度要大于3;
6、在任意连续的10bp中不超过3个SNP
7、去除趋同进化的SNP等; 5.6 系统发育树构建
1、定义:进化树分为分子树与系统发育树。分子树是依据分子数据构建的反映分子系统发育的树。系统发育树就是描述这一群有机体发生或进化顺序的拓朴结构。
2、构建方法
(1)序列类似性比较,主要是基于氨基酸相对突变率矩阵,例如PAM250矩阵,计算不同序列差异性积分作为它们的差异性量度;
(2)通过蛋白质结构比较包括刚体结构叠合和多结构特征比较等方法建立结构进化树。
3、多序列比对构建系统发育树
(1)coregene
(2)MLST
(3)MLVA
(4)全基因组SNP
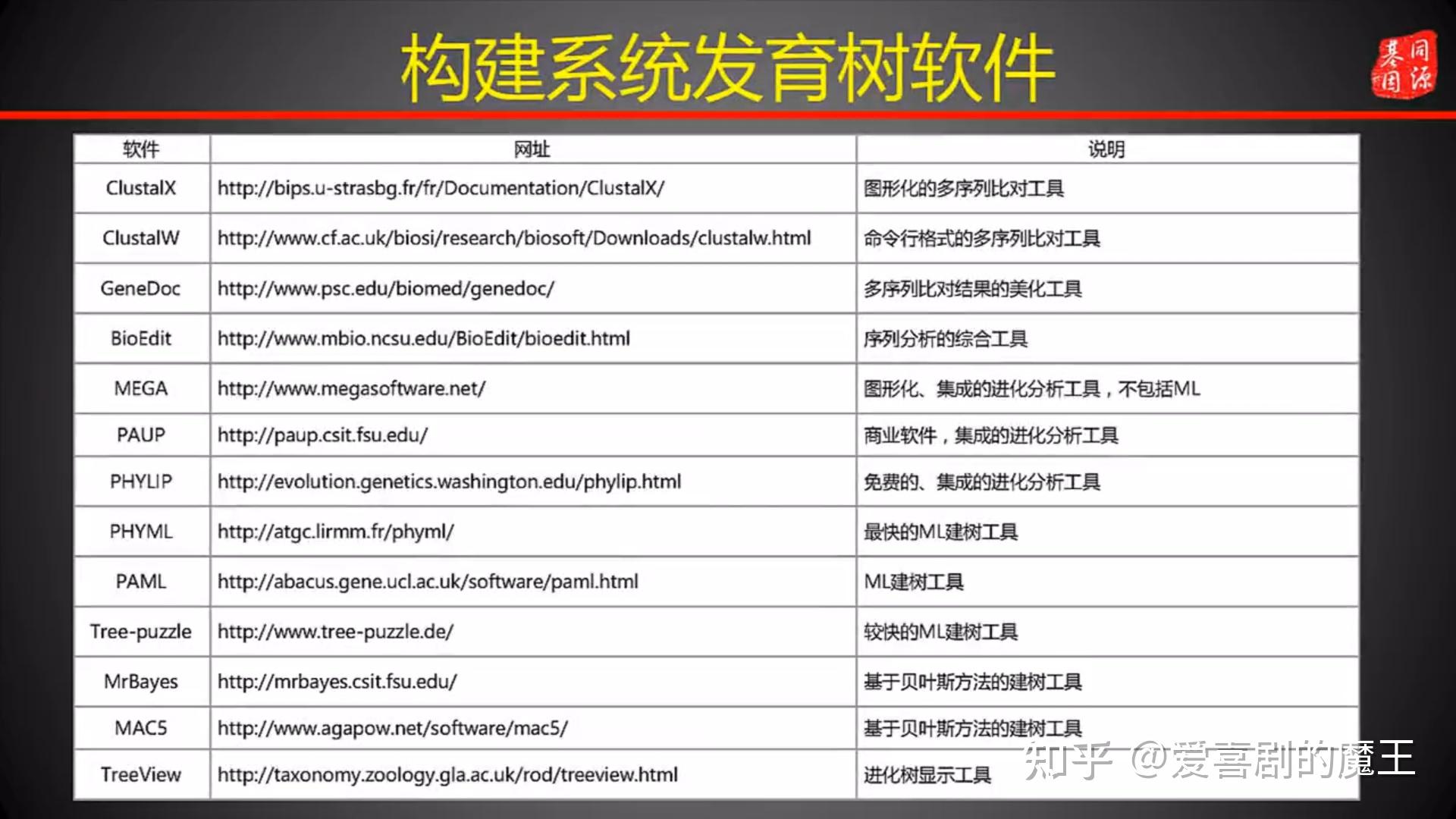
图5.5 构建系统发育树的软件
六、其他
6.1 数据下载
1、常见下载
(1)生物数据分析软件:MyBioSoftware:https://mybiosoftware.com
(2)下载数据库:Nucleic Acids Research
(3)NCBI下载:National Center for Biotechnology Information
2、从NCBI下载数据
视频中的例子太老了,先放一个后面看再完善:爱喜剧的魔王:生物信息学入门——NCBI SRA入门
原文地址:https://zhuanlan.zhihu.com/p/710826840 |
|

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 






 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 发表于 2025-2-20 13:35
发表于 2025-2-20 13:35



