金桔
金币
威望
贡献
回帖0
精华
在线时间 小时
|
登陆有奖并可浏览互动!
您需要 登录 才可以下载或查看,没有账号?立即注册


×
Human Genomics 2022 July
Mamoon Rashid 沙特本阿卜杜勒阿齐兹国王健康科学大学 (KSAU-HS)
# 来自土豪城市的综述 :) 。贵在框架梳理和全景描述很清晰,适合强迫症人群阅读。
## 这篇文章的信息略显滞后(毕竟一年内天翻地覆的,但相比下面2021年的综述,文章的深度和厚度突飞猛进了)
摘要
基因组学正在向数据驱动的科学发展。随着人类基因组学中高通量数据生成技术的出现,我们被大量的基因组数据所淹没。为了从这些基因组数据中提取知识和模式,人工智能尤其是深度学习方法发挥了重要作用。在当前的综述中,我们讨论了深度学习方法/模型在人类基因组学不同子领域的开发和应用。我们通过深度学习技术评估了基因组学的过度和不足领域。基因组工具背后的深度学习算法已在本综述的后面部分进行了简要讨论。最后,我们简要讨论了深度学习工具在基因组中的后期应用。总之,这篇综述对于生物技术或基因组科学家的why, when and how的问题(为啥要用,啥时候用,怎么用的问题)。
前言
了解不同物种的基因组,特别是对超过 30 亿个智人 DNA 碱基对的检查,是基因组研究的一个重要目标。基因组学是一个综合性的视角,涉及生物体内的所有基因,包括蛋白质编码基因、RNA基因、顺式和反式元件等。它是一门数据驱动的科学,涉及下一代高通量技术测序 (NGS),可生成生物体的完整 DNA 数据。这些技术包括全基因组测序 (WGS)、全外显子组测序 (WES)、转录组和蛋白质组分析 [ 1 , 2 , 3 , 4 , 5]。随着最近这些组学数据的快速积累,人们越来越关注生物信息学和机器学习 (ML) 工具,这些工具在多个基因组学实施中具有卓越的性能 [ 6 ]。这些实施包括寻找基因型-表型相关性、生物标志物鉴定和基因功能预测,以及绘制生物医学活性基因组区域,例如转录增强子 [ 7 , 8 , 9 , 10 ]。
机器学习 (ML) 一直被认为是人工智能 (AI) 的核心技术,它可以使用算法并基于数据学习做出关键预测,而不仅仅是遵循指令。具有广泛的技术应用;然而,标准的机器学习方法过于狭窄,无法处理复杂、自然、高维的原始数据,例如基因组学。相反,深度学习 (DL) 方法是目前在基因组学中使用的一个有前途且令人兴奋的领域。它是一种 ML 扩展,通过自动应用神经网络 ( NN )来提取特征[ 11、12、13、14]。深度学习已在图像识别、音频分类、自然语言处理、在线网络工具、自动聊天和机器人等领域得到有效应用。在这方面,将 DL 作为基因组方法论的使用完全易于分析大量数据。虽然仍处于起步阶段,但基因组学中的 DL 有望更新临床遗传学和功能基因组学等领域 [ 15 ]。毫无疑问,DL 算法已经主导了计算建模方法,在这些方法中,它们目前定期扩展以报告各种基因组学问题,包括了解突变对蛋白质-RNA 结合的影响 [ 16 ]、对变异和基因进行优先度排序、诊断患有罕见遗传疾病的患者[ 17],从组蛋白修饰数据中预测基因表达水平 [ 18 ] 并识别与性状相关的单核苷酸多态性 (SNP) [ 19 ]。
虽然 DL 理论的第一个概念起源于 1980 年代,是基于感知器模型和神经元概念 [ 20 ],但在过去十年中,DL 算法已成为大数据的最新预测技术 [ 21 , 22、23]。_ DL 预测模型在基因组学中的最初有效实施是在 2000 年代(图 1)[ 24]。与 DL 模型需要训练大量训练数据集和对强大计算资源的需求相关的困难限制了它们的应用,直到引入现代硬件,例如具有等效结构的高效图形处理单元 (GPU)。现在,如前所述,DL 模型(也称为 DNN)的架构已在不同领域实现。经典的神经网络只有两到三个隐藏层;但是,DL 网络将其扩展到 200 层。因此,“深”这个词反映了信息通过的层数。然而,DL 需要卓越的硬件和大量的并行性才能适用 [ 25]。由于不堪重负的硬件限制和资源需求,人们开发了几个 DL 包和资源以帮助实现DL 模型(在 基因组学的深度学习资源一节中讨论)。
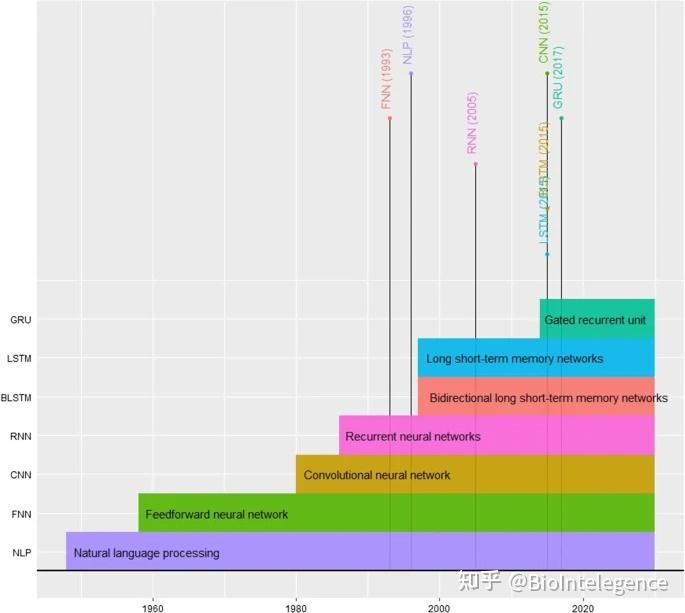
历史沿革
基因组学中软件、硬件 (GPU) 和大数据的发展促进了基于深度学习的预测模型的开发,用于预测基因组中的功能元件。这些来自 NGS 数据的遗传变异预测基因组 DNA 中的剪接位点,通过分类任务预测转录因子结合位点 ( TFBS ) ,对错义突变的致病性进行分类,并预测药物反应和协同作用[ 26、27、28、29、30、31]。增强 DL 实施的技术演进的一个例子是云平台,它提供 GPU 资源作为 DL 解决方案。GPU 可以显着提高训练速度,因为神经网络训练风格可以在某些模型架构情况下更具适应性,从而通过使用更大的处理单元数量和高内存容量来实现快速的数学处理。云计算平台的主要示例包括 Amazon Web Services、Google Compute Engine 和 Microsoft Azure。然而,这些说明仍然需要用户实施模型代码 [ 32 ]。
对于所有 ML 模型,评估指标(evaluation metrics)对于理解模型性能至关重要。基本上,如果基因组数据集自然生成高度不平衡的类,这使得它们需要被 ML 和 DL 模型应用,这些指标是至关重要的。通常在这种情况下应用足够数量的解决方案,例如迁移学习 [ 33 ] 和马修斯相关系数 (MCC) [ 34 ]]。在常识中,每个 ML 任务都可以分为regression task 回归任务(例如预测疾病的某些结果/影响)或classification task分类任务(例如预测疾病的存在/不存在);此外,从这些任务中获得了多个测量指标。通常,基于 ML 回归的方法中使用的一些(但不是全部)性能指标包括:平均绝对误差 (MAE)、均方误差 (MSE)、均方根误差 (RMSE) 和确定系数 (R^2)。相比之下,基于 ML 分类的方法中的性能指标包括:准确度、混淆矩阵、曲线下面积 (AUC) 或/和接收者操作特征下的面积 (AUROC) 和 F1 分数。分类任务最常应用于基因组学研究领域的问题以及比较不同模型的性能。例如,AUC 是评估模型性能的最广泛使用的指标,范围为 [0, 1]。它测量真阳性率 (TPR) 或敏感性、真阴性率 (TNR) 或特异性和假阳性率 (FPR)。此外,F1-score 用于测试高度不平衡数据集中的模型准确度,是准确率和召回率之间的调和平均值(范围也为 [0, 1])。对于 AUC 和 F1 分数,更大的值反映了更好的模型性能 [35、36]。更好地理解 ML 评估指标——目的、计算等——推荐的论文包括 Handelman 等人。(2019)和英格兰和程(2019)。
本文基于深度学习工具/方法在人类基因组学中的当前应用,对其进行了回顾。我们首先在五个主要基因组学领域收集最近(即发表于 2015-2020 年)的 DL 工具:变异调用和注释、疾病变异、基因表达和调控、表观基因组学和药物基因组学。然后,我们简要讨论了基于深度学习基因组学的算法及其应用策略和数据结构。最后,我们提到了基于深度学习的实用资源,以促进深度学习的采用,这对生物医学研究人员和从事人类基因组学工作的科学家来说非常有益。有关基因组学中DL应用领域的更多信息,我们建议:[ 37、38、39]。
基因组学中的深度学习工具/软件/管道
多个基因组学科(例如变异调用和注释、疾病变异预测、基因表达和调控、表观基因组学和药物基因组学)利用生成高通量数据和利用深度学习算法的力量进行复杂预测(图 2)。DNA/RNA 测序技术和机器学习算法,尤其是深度学习的现代发展开启了研究的新篇章,能够将生物大数据转化为基因组学所有子领域的新知识或新发现。以下部分将讨论在各个基因组学领域使用深度学习算法开发的最新软件/工具/管道。
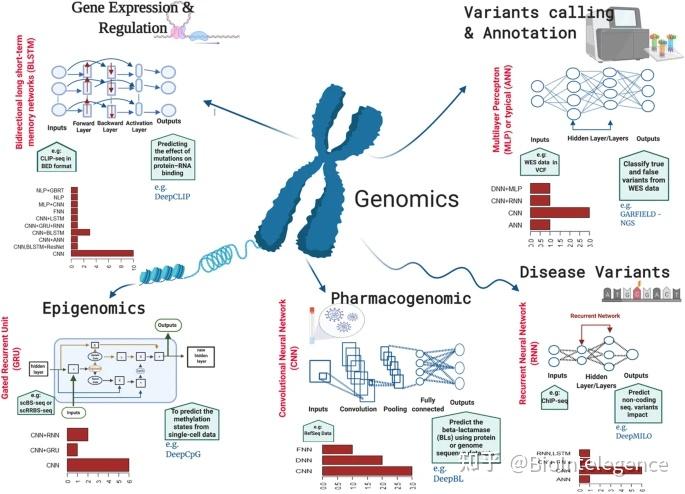
基因组学中的深度学习应用。该图代表了深度学习工具在基因组学的五个主要子领域中的应用。已经为每个基因组子区域展示了一个示例深度学习工具和底层网络架构,并简要提到了它的输入数据类型和预测输出。每个条形图描绘了在该基因组学子领域中作为深度学习工具基础的最常用深度学习算法的频率
突变分析和注释
第一部分讨论最新的 DL 算法在突变分析和注释中的应用。我们提供了用于突变分析和注释的工具/算法的简短列表及其源代码链接(如果可用)(表1),以方便为特定数据类型选择最合适的 DL 工具。
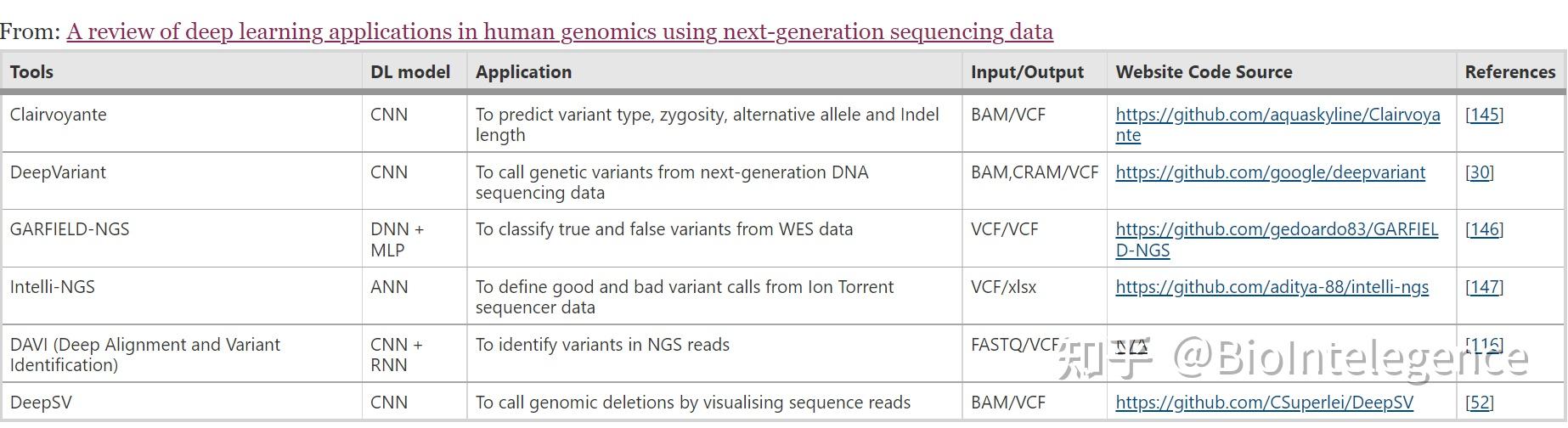
基于深度学习架构的基因组工具/算法,用于突变分析和注释
NGS,包括全基因组或外显子组,为个性化医疗的早期发展奠定了基础,同时它在孟德尔病研究中的已知意义也随之而来。随着大规模并行、高通量测序的出现,对数千个人类基因组进行测序以识别遗传变异已成为基因组学(包括癌症研究)的常规实践。复杂的生物信息学和统计框架可用于变异调用。
高通量测序程序的弱点表现为技术和生物信息学错误率高的吓人 [ 40 , 41 , 42 ]。由于大量的中等或低覆盖基因组序列、短读片段和个体之间的遗传变异,已经产生了许多计算问题[ 43 ]。这些弱点使得 NGS 数据依赖于生物信息学工具进行数据解释。例如,几种变异调用工具广泛用于临床基因组变异分析,例如基因组分析工具包 (GATK) [ 44 ]、SAMtools [ 45 ]、Freebayes [ 46 ] 和 Torrent Variant Caller (TVC; [ 47 ]])。然而,尽管可以进行全基因组测序,但仍有一些实际变体尚未被发现 [ 48 ]。
在下一代测序领域已经提出了当代深度学习工具,以克服传统解释管道的局限性。例如,库马兰等人证明将基于深度学习的变体调用者 DeepVariant 与传统的变体调用者(例如 SAMtools 和 GATK)相结合提高了单核苷酸变体和 Indel 检测的准确度得分 [ 49]。在 DNA 测序数据解释中实施深度学习算法尚处于起步阶段,如最近由 Google 开发的开创性示例 DeepVariant 所示。DeepVariant 依靠输入图像中的图形差异来执行从 NGS 短读长中调用遗传变异的分类任务。它将映射的测序数据集视为图像,并将变体调用转换为图像分类任务 [ 30 ]。但是,此模型不提供有关变异信息的详细信息,例如,确切的替代等位基因和变异类型。因此,它被归类为不完整的变体调用者模型 [ 50 ]。
(译者:个人观点这个办法不是很好, 突变分析需要清晰的知道误差所在,这个领域深度学习的效果不见得优于GATK对实验误差深思熟虑后的规则式的统计检验和过滤。看起来google没有在相关领域再投入时间和精力。看官请自行判断)
后来,引入了几种用于变体调用和注释的 DL 模型。例如,蔡等人。(2019 年)引入了 DeepSV,这是一种遗传变异调用程序,旨在预测从测序读取图像中提取的长基因组缺失(> 50 bp),但不能预测其他类型的结构变异,例如长插入或倒位。它处理 BAM 格式或 VCF 文件作为输入,并以 VCF 形式输出结果。在评估 DeepSV 方面,它与另外 8 个删除调用工具和一个名为 Concod [ 51 ] 的基于机器学习的工具进行了比较。结果表明,尽管 Concod 在训练样本较少的情况下训练时间更短,但 DeepSV 使用相同的数据集显示出更高的准确度得分和更少的训练损失 [ 52]。另一种基因组变异过滤工具 GARFIELD-NGS 可以直接应用于变异调用者输出。它依赖于 MLP 算法来研究从 Ion Torrent 和 Illumina 平台生成的外显子组测序数据集中的真假变体。它通过处理标准 VCF 文件,在低覆盖率数据(高达 30X)下表现出强大的性能,从而产生另一个 VCF 文件。拉瓦西奥等人。(2018 年)观察到,在应用规范管道对疾病相关数据进行变异优先级排序后,GARFIELD-NGS 模型记录的错误候选变异显着减少 [ 53 ]。
后来 Clairvoyante 模型被引入来预测变异类型(SNP 或 Indel)、杂合性zygosity、等位基因变化allele alternative和 Indel 长度。因此,它克服了 DeepVariant 模型缺乏完整变异细节的缺点,包括精确的替代等位基因和变异类型。Clairvoyante 模型专门设计用于利用 SMS 技术(例如 PacBio 和 ONT)生成的长读长测序数据,尽管它通常也适用于短读长数据集 [ 50]。
(译者注:
该工具已经迭代进入 Clair3 GitHub - HKU-BAL/Clair3: Clair3 - Symphonizing pileup and full-alignment for high-performance long-read variant calling
这个工具包做的非常好,建议刚接触的朋友可以了解一下,很适合后续工作时触类旁通。
文章见 https://www.biorxiv.org/content/10.1101/2021.12.29.474431v1)Singh 和 Bhatia (2019) 引入了另一个变体调用者和注释模型 Intelli-NGS。一种变体调用基于人工神经网络 (ANN),它利用 Ion Torrent 平台生成的数据来有效识别真假。Intelli-NGS 将任意数量的 VCF 文件作为批处理输入并按顺序处理它们。处理后的数据会生成一个与每个 VCF 文件相关的 Excel 表,其中包含所有变体的 HGVS 代码 [ 54 ]。总而言之,几项研究证实了深度学习在遗传变异调用和测序数据注释方面的能力。
疾病变体
表2列出了基于深度学习的致病变异预测模型、它们的应用和带有源代码(如果有)的输入/输出格式。
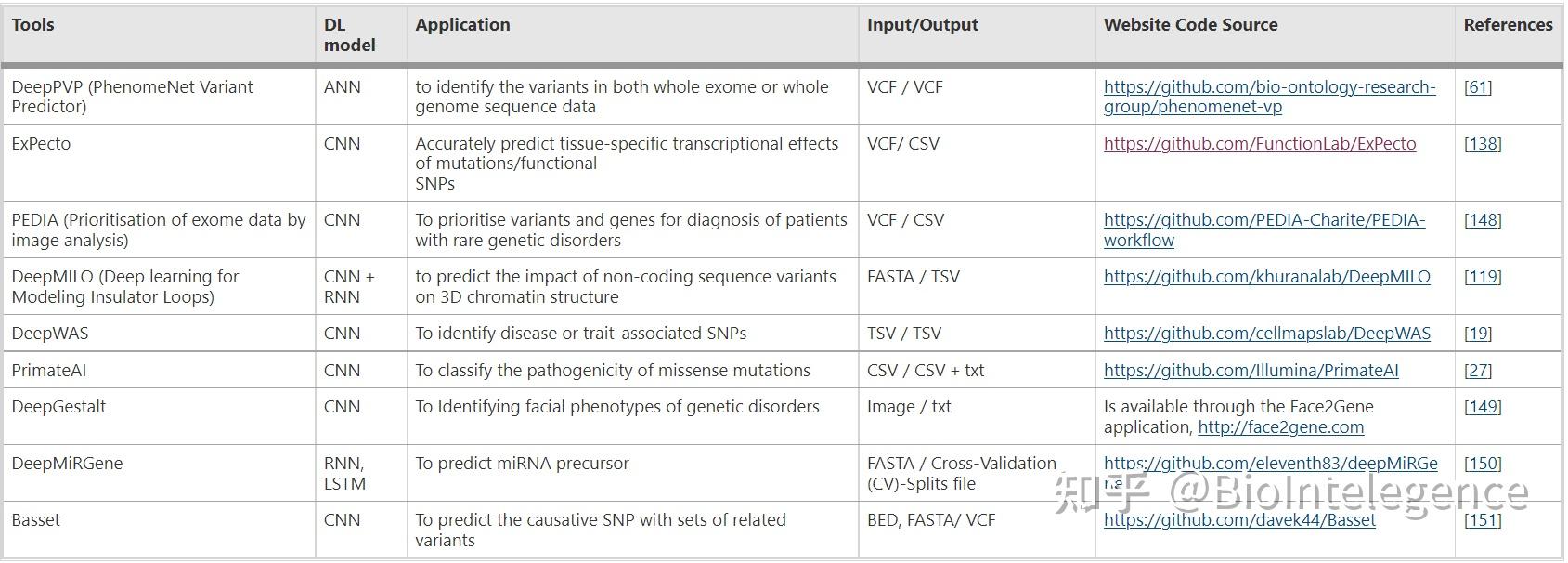
基于深度学习架构的疾病变异基因组工具/算法
考虑到来自患者亲属或相关队列的额外数据,医学遗传学家经常在变异调用和注释后对观察到的遗传变异进行优先排序和过滤(Müller 等人 [ 55 ])。变异优先级是一种在基因筛查中确定最可能的致病变异的方法,该变异会损害基因功能并作为疾病表型的基础 [ 56 ]。变异优先级涉及变异注释以发现临床上不重要的变异,例如同义、深内含子变异和良性多态性。随后,剩余的变体,例如已知变体或临床意义未知的变体 (VUS),变得可实现 [ 57]。此外,例如,解释个体罕见遗传变异的并发症以及了解它们对疾病风险的影响会影响诊断测序的临床能力。例如,罕见遗传病中大量且不常见的 VUS 代表了个性化医疗和健康人群评估的测序实施中的一个具有挑战性的障碍(Sundaram 等人,2018 年)。尽管 GWAS 等统计方法在将遗传变异与疾病相结合方面取得了巨大成功,但它们仍然需要大量抽样来区分罕见的遗传变异,并且无法提供有关从头变异的信息 (Fu et al., 2014)。因此,当前的注释方法,如 PolyPhen [ 58 ]、SIFT [ 59 ] 和 GERP [ 60],尽管面临一些缺点,但代表了对致病变异进行优先排序的有益方法。对于这些问题,已经实现了基于 DL 的模型,以启用一种强大的方法来利用深度神经网络 (DNN) 架构对变体进行优先级排序,例如 Basset 模型,一种变体注释器,它依赖于 CNN 算法并被设计为利用 DNase I 超敏反应测序数据作为输入预测致病 SNP(Kelley、Snoek 和 Rinn,2016)。
临床和分子验证不能被计算机预测模型取代;然而,从某种意义上说,它们可以有助于减少等待结果的时间,并可以优先考虑变体以进行进一步的功能分析。这些可预测的模型主要适用于几个知之甚少的候选变体传达某些表型时 [ 27]。随着 NGS 技术的提出,医学遗传学发生了重大变化,尤其是 WGS,因为它能够解释整个人类基因组中编码和非编码片段的基因组变异。最近,有几种基于机器学习的方法可以优先考虑非编码变体。尽管如此,识别癌症等复杂性状中与疾病相关的变异仍然具有挑战性。此外,需要与某种表型相关的大多数阳性变体来预测一般和精确的新相关性(Schubach 等人,2017)。最近,已经提出了几种 DL 方法来克服这些挑战。例如,DeepWAS 模型依赖于 CNN 算法,该算法允许预测每个变体对众多细胞类型特异性染色质特征的监管影响。DeepWAS 模型的关键结果是直接确定与疾病相关的 SNP,这些 SNP 对相关组织中的某些染色质性状具有共同影响。DeepWAS 模型展示了在结合各种资源和组织的表达和甲基化数量性状基因座数据(分别为 eQTL 和 meQTL)后检测与疾病相关的、转录活跃的基因组位置的能力。19 ]。然而,一些深度学习算法已被描述为发现新基因。出于这个原因,深度学习方法特别适合对尚未与特定疾病表型相关的基因进行变异研究 [ 61 , 62 ]。
(译者注:
请允许我多一嘴,和上面的突变分析variant-calling这个生信的看家本领不同,
疾病相关的突变确定是个生物学上和临床上都非常tricky的问题。在可见的未来随着数据的积累这个问题会更加凸显。
用户只能福尔摩斯式地利用工具文献阅读的经验和突变频率及病人中的分布来反复观察,得到一个有用的突变通常就是能发一篇case。
这个问题的解决,可能不只是算法上改善或者调参,而是需要借助NLP的工具对疾病术语和已有的文献报道进行归纳,
解决方案显得非常tedious而收获不会太大。可是这是生物学假设中的核心问题,是非常难但是意义非常重大的课题。
初学者建议不要单打独斗的挑战这个领域。需要比现有工具更强大的人工智能帮手。
好处是大家做WGS的越来越多,可能会促进这个方向的技术进步。 请继续读。谢谢~
)基因表达与调控
在本节中,我们专注于基因组中基因表达和调控领域最有效的基于深度学习的工具。我们列出了几个应用各种深度学习算法的模型,并总结了主要在剪接和基因表达应用中的信息和源代码(如果有)(表3)。
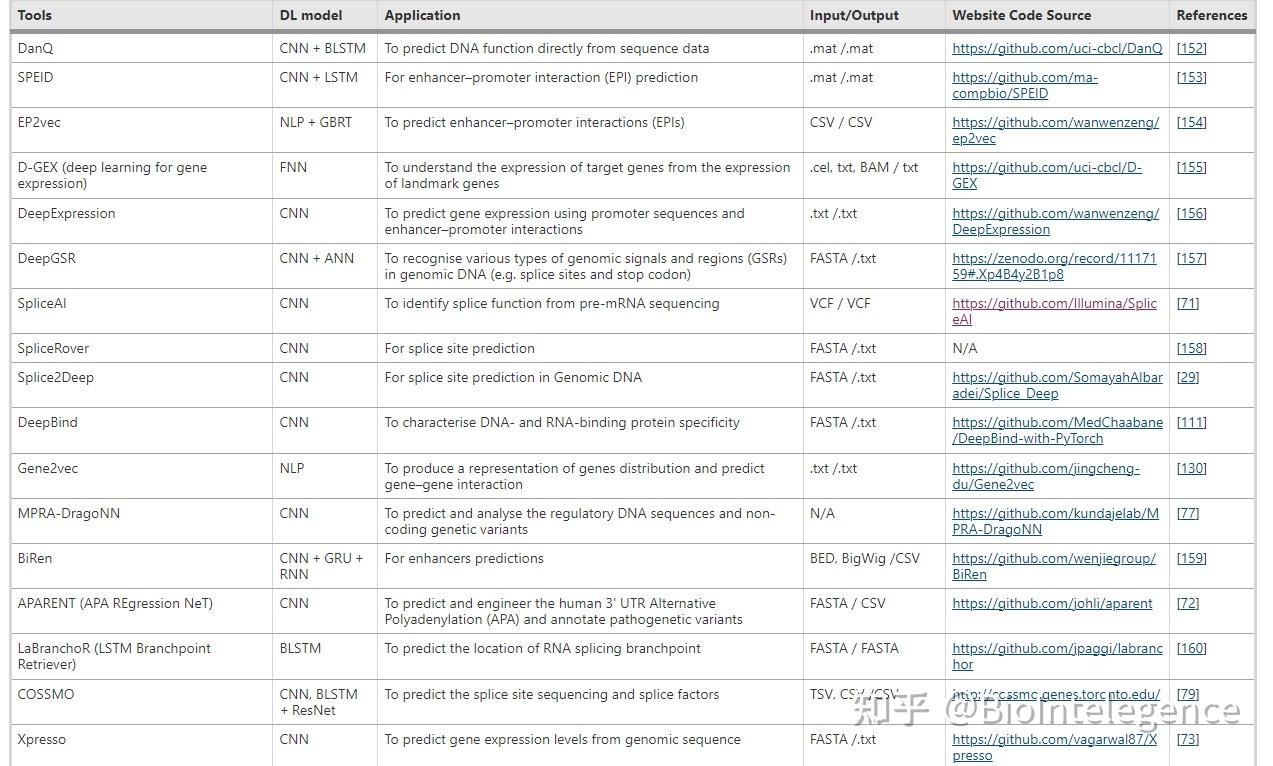
这个方向显然是比较低垂的果实,你看搞了这么多工具出来。
基因表达涉及到功能性蛋白质生产的初始转录调节因子(例如前 mRNA 剪接、转录和多腺苷酸化)[ 63 ]。测试数千个合成序列的高通量筛选技术提供了有关基因表达定量调控的丰富知识,尽管存在一些局限性。主要限制是无法使用实验或计算技术探索巨大的生物序列区域 [ 64]。尽管最近的 NGS 技术在基因调控领域提供了丰富的知识,但大多数天然 mRNA 筛选方法仍然利用染色质可及性、ChIP-seq 和 DNase-seq 信息;他们专注于研究启动子区域。因此,需要一种稳健的方法来了解基因调控结构的各个区域及其网络表达连接之间的关系[ 65 ]。同样,目前的 RNA 测序技术已经赋予了单细胞直接测序的能力,被称为单细胞 RNA 测序 (scRNA-seq),它允许以独特的意图查询生物系统。例如,scRNA-seq 的数据为细胞异质性提供了有价值的信息,可以扩展对人类疾病和生物学的解释。66、67]。 scRNA-seq 数据理解的主要应用涉及检测细胞的类型和状态 [ 68 , 69 ]。然而,两个主要的计算问题包括如何对数据进行聚类以及如何检索它们[ 70 ]。
深度学习为构建将调控序列元素与分子表型联系起来的预测方法取得了重大进展 [ 71 , 72 , 73 , 74 ]。就在最近,Gundogdu 和他的同事 (2022) 展示了一个基于深度神经网络 (DNN) 的出色分类模型。它限制了基因之间功能网络的多种先前生物学信息,以了解 scRNA-seq 数据的生物学意义 [ 70 ]。此外,李等人。(2020) 提出了一种 DESC,一种基于 python 实现的无监督深度学习算法,它可以迭代地理解集群特定基因表达的表示和 scRNA-seq 分析集群任务 [75 ]。此外,深度学习模型也已应用于单细胞测序数据。其深度神经网络 (DNN) 模型旨在测量结直肠癌和乳腺癌大量 scRNA-seq 数据中的免疫浸润。这种方法允许量化特定类型的免疫细胞,例如 CD8 + 和 CD4Tmem 以及淋巴细胞的一般群体以及基质含量和 B 细胞 [ 76 ]。
最近,贾加纳坦等人。(2019) 构建了 SpliceAI,这是一个深度残差神经网络,仅使用前 mRNA 转录本测序作为输入来预测剪接功能。一个架构包含一个 32-dilated 卷积层,用于识别序列确定跨越巨大的基因组间隙,因为有数以万计的核苷酸分隔剪接供体和剪接受体 [ 71 ]。
许多实验数据集,例如 ChIP-seq 和 DNase-seq 分析,并不直接测量对基因表达的影响;然而,它们是深度神经网络方法的理想补充。例如,Movva 等人。(2019)引入了基于 CNN 架构的 MPRA-DragoNN 模型,用于预测和分析从 (MPRA) 数据测量的非编码 DNA 测序数据的转录调控活性。在 Sharpr-MPRA 评估中使用的最小启动子或强启动子上游克隆了 295 bp顺式调控元件的 K562 和 HepG2 细胞系中大约 16 K 不同的调控区域[ 77]。由 Agarwal 和 Shendure 引入的一个非常现代的 DL 模型,命名为 Xpresso 模型,一种深度卷积神经网络 (CNN),联合对启动子序列及其相关 mRNA 稳定性特征进行建模,以预测 mRNA 的基因表达水平。有趣的是,Xpresso 模型很容易在多种任意细胞类型上进行训练,即使它们缺乏实验信息,例如 ChIP 和 DNase [ 73 ]。张Z.等。(2019) 开发了一种基于深度学习的模型,称为 DARTS;深度学习增强了转录剪接的 RNA-seq 分析,即使用各种可变剪接的广泛 RNA-seq 资源。它由两个主要模块组成:深度神经网络 (DNN) 和贝叶斯假设检验 (BHT) [ 78]。Bretschneider 等人设计的更多基于 DL 的模型(特别是四种不同的 CNN 架构)。(2018),命名竞争剪接位点模型(COSSMO),它适应各种数量的选择性剪接位点,并通过全基因组交叉验证精确估计它们。该框架由卷积层、通信层、长短期记忆 (LSTM) 和残差网络组成,相应地,用于从 DNA 序列中发现相关基序。在每个假定的剪接位点中,使用的模型输入是 DNA 和 RNA 序列,在选择性剪接位点和相反的组成型剪接位点周围有 80 个核苷酸宽的窗口以及内含子长度。该模型的输出是对每个假定剪接位点的百分比选择指数 (PSI) 分布的预测。COSSMO模型的所有性能均超过MaxEntScan;然而,四个框架之间存在很大的性能差异,其中循环 LSTM 在通信网络上达到了最佳精度,没有考虑拼接点排序 [79 ]。然而,为了在不完美的生物情况下学习异构数据集之间的自动化关系,深度学习模型提供了前所未有的机会。
(译者注:啥也不说了 这个方向有嗷嗷多的数据可以用,孵育了嗷嗷多的文章)
建议初学者从这里入手。再搞个三五年,一点问题木有。
表观基因组学
本节讨论了一些表观基因组学挑战,并总结了表观基因组学中最新的深度学习模型、它们的实现、数据类型和源代码(表4)。不基于基因型修饰的表型修饰称为表观遗传学。它被定义为研究基因表达中的可遗传修饰,其中不包括 DNA 序列变化[ 80]。表观基因组机制,包括 DNA 甲基化、组蛋白修饰和非编码 RNA,被认为是了解疾病发展和寻找新治疗靶点的基础。尽管在临床实施中,表观遗传学尚未完全应用。最近,在开发数据解释工具以产生表观遗传数据的下一代测序和微阵列技术的进步方面出现了进展。尽管在体内发生了几种标记相互作用和基因型[ 81]。之前的几项研究已经揭示了深度学习模型在表观基因组学中的基本应用。他们在基于 DNase-Seq 数据预测 3D 染色质相互作用、单细胞数据集和组蛋白修饰位点的甲基化状态方面取得了非常多的成果 [ 62 , 82 , 83 , 84 ]。
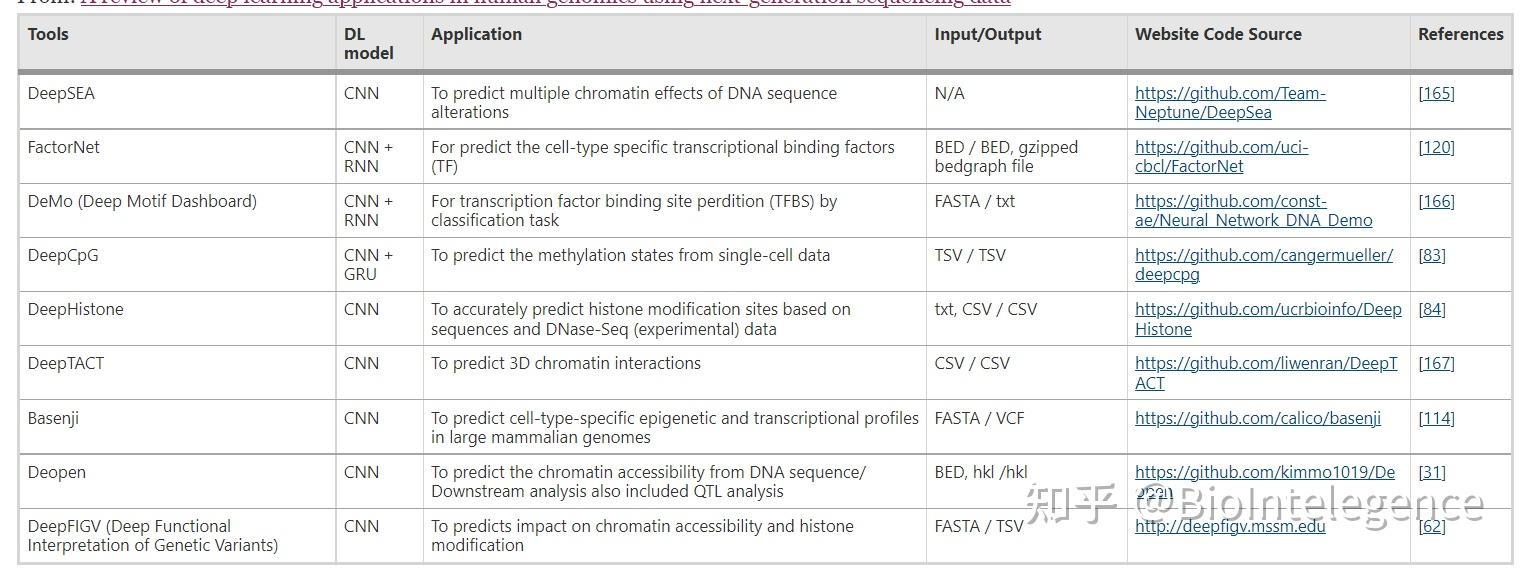
刘等人。(2018 年)引入了一种混合深度 CNN 模型 Deopen,该模型用于根据学习的调控 DNA 序列代码预测整个基因组内的染色质可及性。为了分析评估 Deopen 在捕获基因组可访问性代码方面的功能,从二进制分类的角度进行了一系列实验 [ 31 ]。作为 Deopen 应用的一个例子,在对雄激素敏感的人类前列腺腺癌细胞系 (LN-CaP) 中,Deopen 模型恢复的 EGR1 被认为在前列腺癌基因治疗中作为治疗靶点发挥了关键作用 [ 31 , 85]。最近,尹等人。(2019) 提出了 DeepHistone 框架,这是一种基于 CNN 的算法,用于预测对各种特定位点标记的组蛋白修饰。为了进行精确预测,该模型将 DNA 序列数据与染色质可及性信息相结合。它揭示了区分功能性 SNP 与其相邻遗传变体的能力,因此有可能用于研究假定的疾病相关变体的功能影响 [ 84 ]。因此,基因组研究需要有效的深度学习模型来阐明表观基因组修饰对下游输出的影响。
药物基因组学
我们列出了最深思熟虑的深度学习药物基因组学模型、它们的常见用途、输入/输出格式和代码源(表5)。尽管在过去几年中人们对深度学习方法产生了极大的兴趣,但直到最近,深度学习工具还很少用于药物基因组学问题,例如预测药物反应 [ 86 ]。关于从巨大基因簇到全基因组的遗传变异与不同药物的影响之间关联的知识被称为药物基因组学 [ 87]。现代治疗方法的一个关键挑战是了解变异的潜在机制。有时,某个人群的药物反应分布显然是双峰的,这表明一个变量具有主导功能,通常是遗传的。尽管如此,了解药代动力学或药效学的潜在机制可用于检测候选基因,其中这些基因变体的功能可以解释各种药物反应(88)。临床实验在研究药物组合效率的过程中会产生各种错误,这既费时又费钱。此外,它可能使患者接受过度危险的治疗 [ 89 , 90]。为了在不伤害患者的情况下确定替代药物协同策略,使用了几种浓度的几种药物用于癌细胞系的高通量筛选 (HTS) [ 91 ]。利用现有的 HTS 协同数据集允许使用精确的计算模型来研究巨大的协同空间。这种可靠的模型将为体外和体内研究提供方向,它们是朝着个性化医疗迈出的重要一步,例如,抗癌协同预测方法、系统生物学 [ 92 ]、动力学方法 [ 93 ] 和基于计算机的模型单药和剂量反应治疗后的基因表达筛选[ 94]。尽管如此,这些方法仅限于特定的靶点、通路或某些细胞系,有时需要使用特定化合物处理的细胞系的特定组学数据集 [ 95 ]。
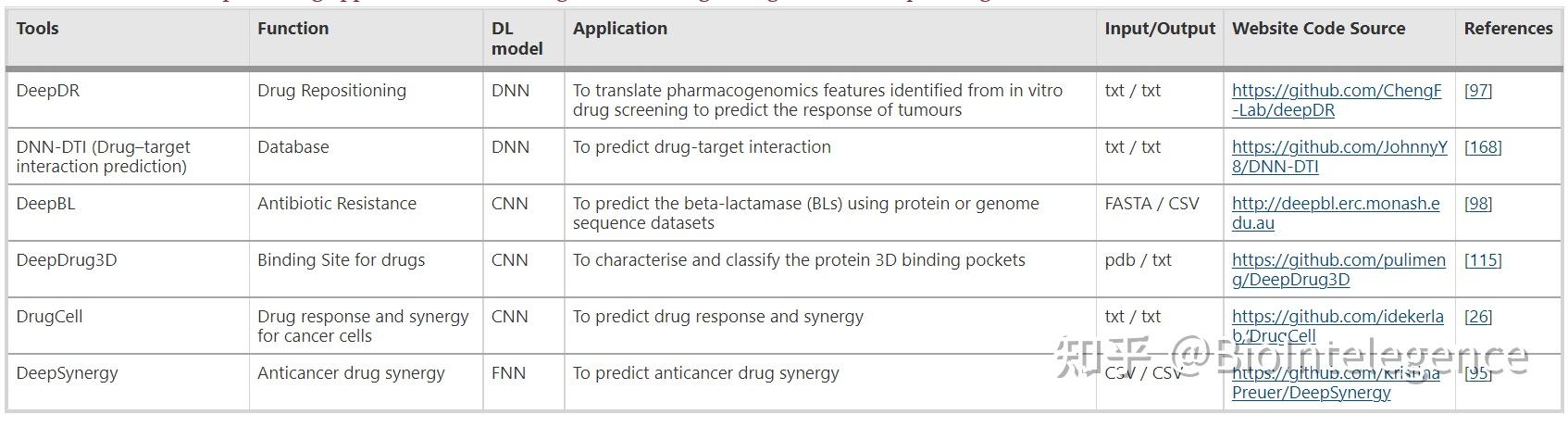
为了研究这些药物基因组学关联,使用了统计数据,例如方差分析 (ANOVA) 检验。例如,这可以识别患者体内发生的致癌变化,这些变化是细胞系中药物敏感性差异的指标。为了超越药物与实际药物反应预测的关系,可以采用多种统计和机器学习方法,从线性回归模型到非线性模型,例如核方法、神经网络和 SVM。这些方法的一个主要弱点是大量输入特征以及低样本数,例如在标准基因表达分析中,输入基因(或特征)的总数超过了样本数。克服低采样数问题的最新策略是使用多任务模型 [96].
据报道,深度学习方法非常适合基于细胞系组学数据集的治疗反应预测任务 [ 95 , 97]。其中一个例子是 DrugCell,一种可视神经网络 (VNN) 解释模型,用于研究人类癌细胞在治疗反应中的结构和功能。它将模型的中心机制与人类细胞生物学结构配对。允许预测任何癌症中的任何药物反应,然后巧妙地计划治疗的有效组合。DrugCell 的开发是为了在一个可解释的模型中捕获治疗反应的两个要素,该模型具有两个部分,即 VNN 整合细胞基因型和人工神经网络 (ANN) 整合药物设计。第一个 VNN 模型输入包括人类细胞中分子子系统之间的层次关联的文本文件,其中包含基因本体 (GO) 数据库中的 2086 个生物过程标准。第二个 ANN 模型输入是传统的 ANN,集成了 Morgan 医学指纹的文本文件,即规范矢量符号的化学结构。这两个部门的输出被组合成单层神经元,产生给定基因型对某种治疗的反应。每种药物的预测准确性分别揭示了具有显着准确性的药物亚群。这反过来又与先前模型中应用的最先进的回归方法竞争,以预测药物反应。此外,将 DrugCell 与仅针对药物设计和标记组织进行训练的并行神经网络模型进行比较,其性能大大优于基于组织的模型。这意味着 DrugCell 已经从体细胞突变中学习到超过仅组织方法的数据 [26 ]。另一个名为 DeepBL 的最新模型基于基于 Small VGGNet 结构(一种 CNN)和 TensorFlow 库执行的深度学习架构。这种方法检测β-内酰胺酶 (BLs) 及其对 β-内酰胺类抗生素产生抗性的品种,并以蛋白质序列作为输入。它基于经过充分解释的海量 RefSeq 数据集,涵盖从 NCBI 数据库中提取的 > 39 K BL。将该模型与其他基于机器学习的传统算法(包括 SVM、RF、NB 和 LR)进行比较,在对包含超过 10 K 序列的独立测试集进行评估后,DeepBL 的表现优于它们 [ 98 ]。直到最近,药物基因组学中的深度学习应用仍在考虑之中。
基因组学中使用的深度学习算法/技术
最近在基因组学部分的深度学习工具/软件/管道中提到的可实现模型的成就表明,深度学习是基因组研究中的一项强大技术。在这里,我们专注于最近在基因组应用中应用的深度学习算法:卷积神经网络 (CNN)、前馈神经网络 (FNN)、自然语言处理 (NLP)、递归神经网络 (RNN)、长短期记忆网络 (LSTM) )、双向长短期记忆网络 (BLSTM) 和门控循环单元 (GRU;表6;图 1 )。
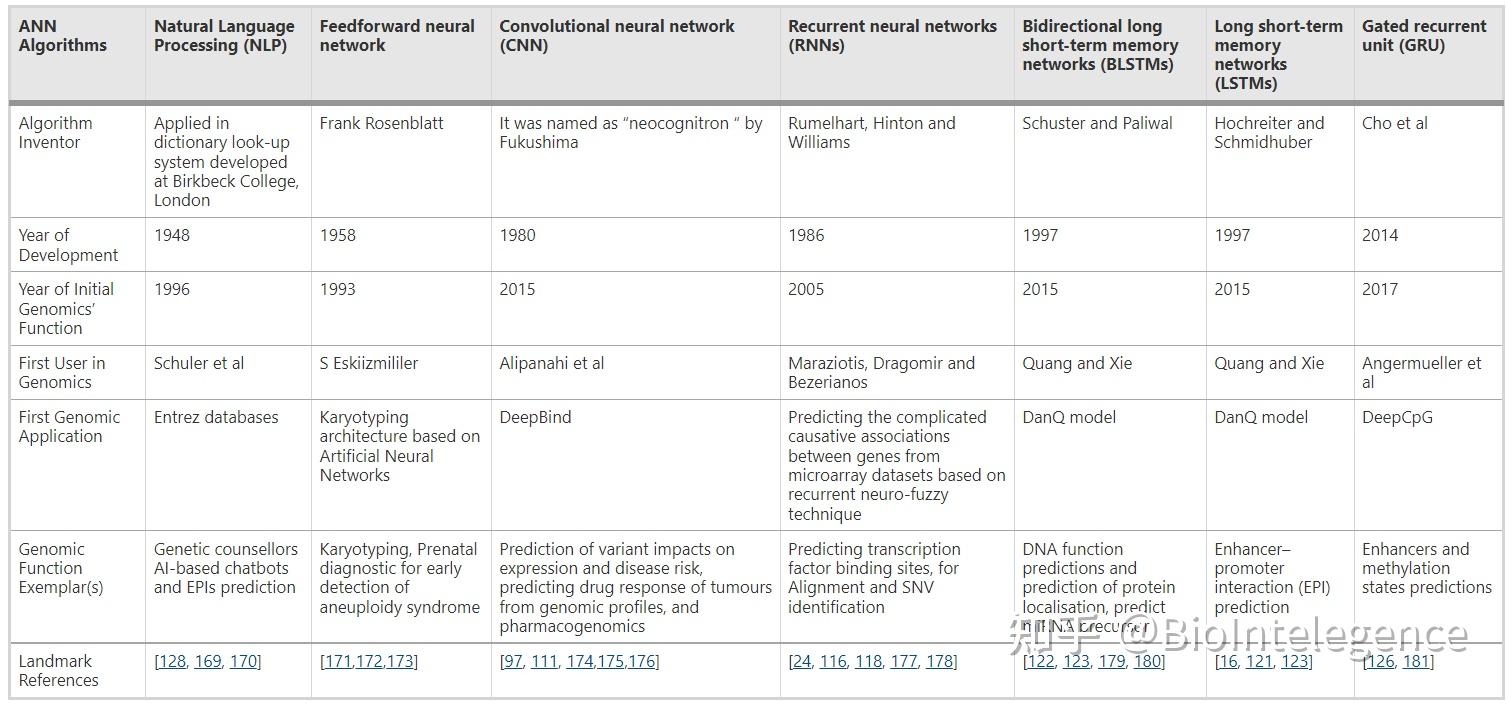
#这部分跳过了,都是作者东抄抄西抄抄的内容。
基因组学的深度学习资源
我们收集了基于深度学习架构开发的最高效、用户友好的基因组资源(表7)。尽管这些工具在基因组学和生物信息学方面取得了巨大成功,但各种深度学习解决方案和模型的采用仍然有限。造成这种情况的一个原因是缺乏基于深度学习的已发布协议来适应需要大量数据工程的新的异构数据集 [ 135]。在基因组学中,高通量数据(例如 WGS、WES、RNA-seq、ChIP-seq 等)用于训练神经网络,并已成为疾病预测或理解调控基因组学的典型。同样,由于缺乏包容性、通用、实用的生物学深度学习库,开发新的深度学习模型和在新数据集上测试当前模型面临着巨大挑战[ 136 ]。在这方面,软件框架和基因组包对于在采用新的研究问题或假设、结合原始数据或使用不同的神经网络结构进行研究方面取得快速进展是必要的 [ 135]。为了促进基因组学中的 DL 模型实施,以下软件包或库可能对基因组科学家和生物医学研究人员至关重要。
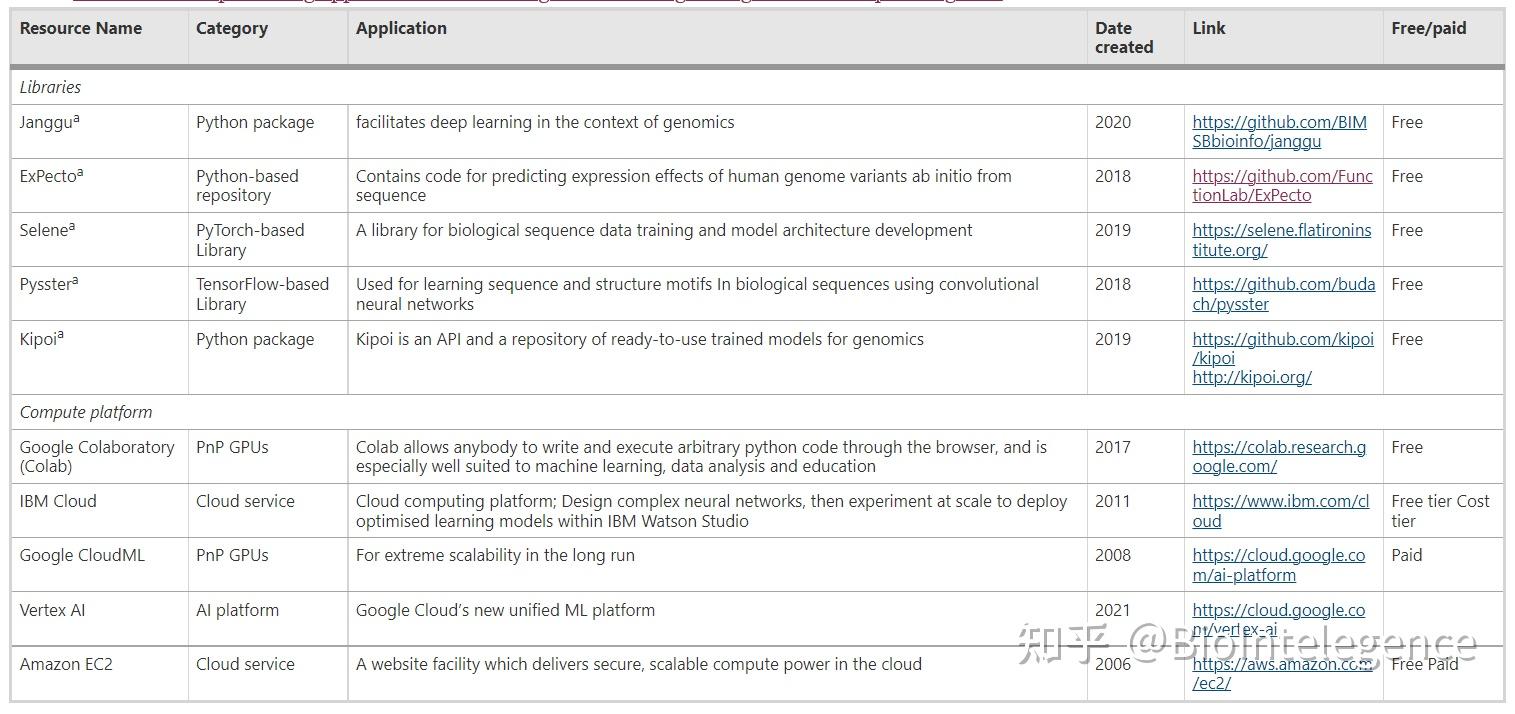
Janggu是一个基于深度 CNN 的深度学习 python 库,用于基因组实现。它旨在通过支持灵活的神经网络原型模型来实现数据采购设施和模型评估。Janggu 库提供了三个用例:转录因子预测、利用和增强已发布的深度学习设计以及预测启动子的 CAGE 标签计数标准化。该库提供了方便的访问和预处理功能,可以将数据从标准文件格式(例如 FASTA、BAM、Bigwig、BED 和narrowPeak)转换为 BigWig 文件 [ 135 ]。
Selene是一个基于 PyTorch 的深度学习库,用于生物序列数据训练和模型架构开发。Selene 支持遗传变异效应的预测,并将变异分数可视化为曼哈顿图。它还从给定的输入数据集自动生成训练、测试和验证拆分。此外,Selene 自动训练数据并可以在测试集上检查模型,从而生成可视化图形来显示模型的性能 [ 137 ]。
ExPecto是一种变体优先排序模型,用于预测启动子近端测序区域的广泛调控区域(~ 40 kb)范围内的基因表达水平。它依赖 CNN 将输入序列转换为表观基因组特征。ExPecto 促进了罕见变异或前所未有的变异预测。这是因为其独特的设计架构,在训练过程中不使用任何变体信息。ExPecto 处理 VCF 文件并输出 CSV 文件 [ 138 ]。
Pysster是一个基于CNN的python库包,用于生物测序数据的训练和分类。Pysster 提供自动超参数优化和主题可视化选项以及它们的位置和类丰富信息 [ 139 ]。
Kipoi(希腊语为“花园”;发音为“kípi”)是一个基因组库,用于共享和重用经过训练的基因组相关模型。Kipoi 提供了来自 22 项不同研究的 2K 多个经过明确训练的模型,涵盖了重要的预测基因组任务。预测包括染色质可及性测定、转录因子结合和 DNA 序列的可变剪接 [ 136 ]。
实施这些深度学习、基于基因组的库/包需要访问计算机能力并熟悉基于 Web 的资源(表7)。几个主要的云计算平台以用户友好的方式提出了按需 GPU 访问,包括 Google CloudML、IBM 云、Vertex AI和Amazon EC2 [ 140、141、142]。这些基于云的机器需要用户配置和安装适当的通用 GPU 编码环境。同时,对于需要避免半手动设置方法的用户,提供专家即插即用(PnP)平台GPU访问,例如Google Colaboratory(Colab)。Google Colab 被认为是最简单的基于 python 的 notebook 替代品,可连续 12 小时免费使用 K80 GPU [ 143、144 ]。表7提供了在基因组学中应用深度学习的资源(包/库和网络平台)的链接。
结论
这份手稿列出了在基因组学的不同子领域开发的不同深度学习工具/软件,以完成各种基因组分析的预测任务。我们详细讨论了不同基因组学分析中的数据类型,以便读者能够初步了解使用人类基因组学数据集开发基于深度学习的预测模型的基本要求。在手稿的后半部分,向基因组科学家简要介绍了不同的深度学习架构,以帮助他们针对特定数据类型和/或问题决定深度学习网络架构。我们还简要讨论了深度学习技术在基因组学中的后期应用及其根本原因和解决方案。在手稿结束时,各种计算资源,提供软件包或库和基于网络的计算平台,作为研究人员利用基因组数据集创建他们的第一个深度学习模型的指针。总之,这项及时的审查有可能帮助基因组科学家采用最先进的深度学习技术来探索基因组 NGS 数据集和分析。这对生物医学和人类基因组学研究人员肯定是有益的。
## 抱歉文章中的表格我没有进行排版,直接用的截图,原文是OA的,请参考开头的链接。
## 很多朋友默默的点了收藏,点个赞瓦,泵油~
原文地址:https://zhuanlan.zhihu.com/p/554271013 |
|

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 






 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 发表于 2025-2-18 17:19
发表于 2025-2-18 17:19



