金桔
金币
威望
贡献
回帖0
精华
在线时间 小时
|
基于k-mers的GWAS分析的模块化工作流程
识别基因型和表型之间的关联是理解复杂生物性状遗传结构的基础。自2000年以来,全基因组关联研究(Genome-wide association studies, GWAS)一直是检测遗传变异和表型之间是否存在关联的首选方法。通过GWAS,对不同样品队列的数千个性状进行了调查,已经产生并报告了许多具有统计学意义的关联。这些发现使我们更好地认知复杂的人类特征和疾病,有助于改善植物育种和动物健康,并在其他方面显著影响着我们对遗传学的理解。
经典的GWAS方法使用全基因组范围的单核苷酸多态性(single- nucleotide polymorphisms, SNPs)作为基因型数据。在一个标准的GWAS分析中,使用统计模型测试SNP标记与表型性状的统计学显著关联。GWAS利用标记和因果变异之间的连锁不平衡(LD)信息来识别性状相关的基因座。然而,尽管这种方法具有明显的优势,但也有各种局限性。当前研究已表明,由于连锁不平衡的特性,GWAS往往无法确定因果变异。此外,这些研究主要依赖SNPs作为遗传变异的标记,而往往忽略其他变异,如结构变异,因此有时只能解释复杂遗传力的某些小的方面,尤其是在涉及高度复杂性状的情况下。此外,GWAS还可以识别假阳性的关联,并且无法捕获由罕见变体引起的关联。
GWAS分析的质量还取决于参考基因组的可用性和质量。传统的GWAS依赖于将测序读数比对到参考基因组,然后检测遗传变体。这个比对步骤可能会在变异检测过程中造成偏差,因为参考基因组通常是不完整的,并且可能无法代表群体内的全部遗传变异。此外,有误的比对可能导致不正确的变体检测,尤其是在复杂的基因组和/或重复区域周围。
由于这些限制,目前科研工作者正在开发更多、准确性更好、更易用的GWAS方法。这些较新的方法包括但不限于使用单个物种的泛基因组而不是单个参考基因组,以及使用SNPs之外的新标记,如结构变异和k-mers。
下面的流程图展示了一种基于k-mers的GWAS分析的模块化工作流程:
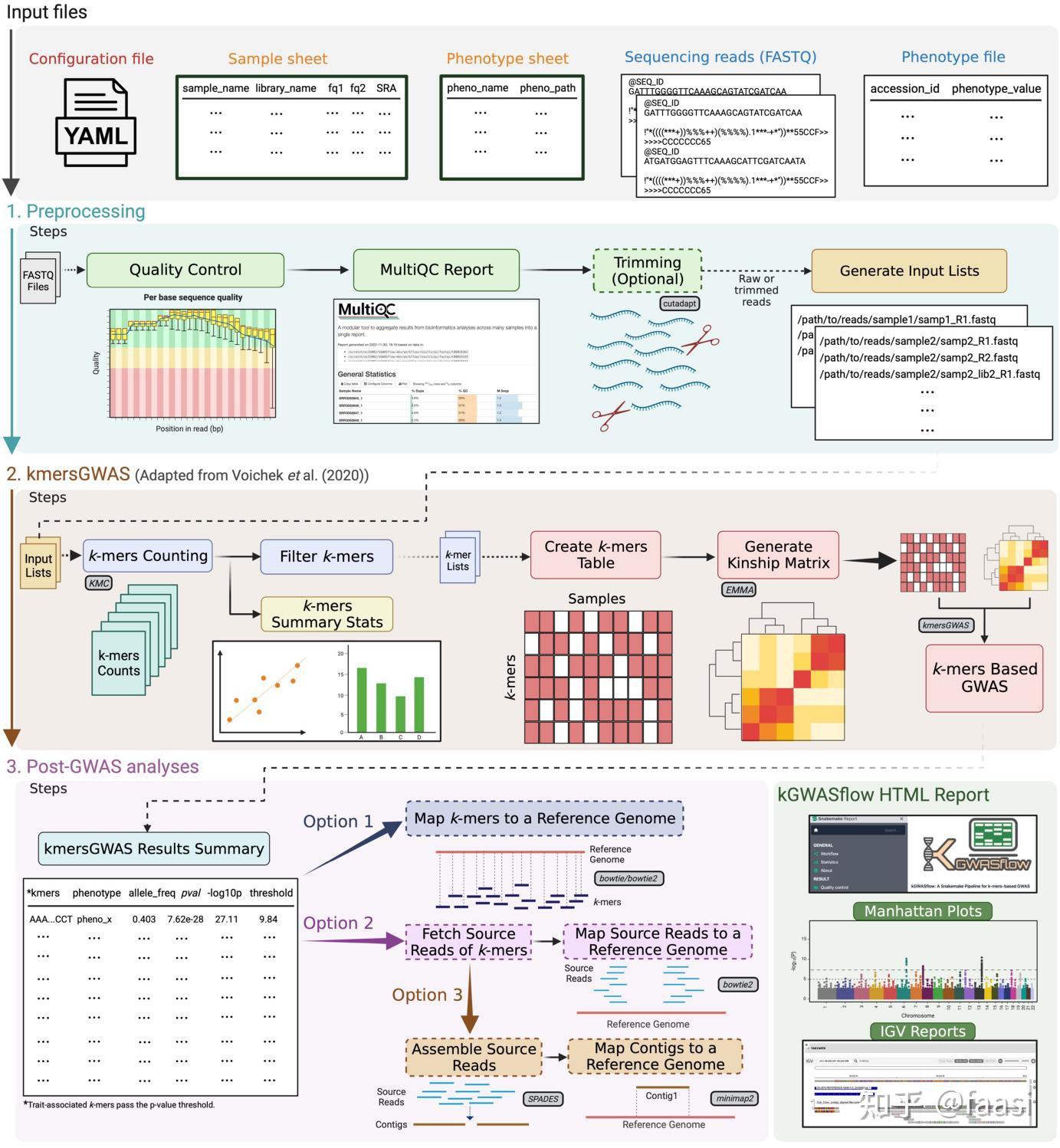
对GWAS分析设计感兴趣的老师或同学可以关注公众号“多组学数据分析和信息挖掘”(微信号:bioInforMiner)与我联系!
Reference
Corut AK, Wallace JG. kGWASflow: a modular, flexible, and reproducible Snakemake workflow for k-mers-based GWAS. G3 (Bethesda). 2023 Dec 29;14(1):jkad246. doi: 10.1093/g3journal/jkad246. PMID: 37976215; PMCID: PMC10755180. |
|

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 






 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 发表于 2025-1-30 19:44
发表于 2025-1-30 19:44


 发表于 2025-1-30 19:44
发表于 2025-1-30 19:44



