金桔
金币
威望
贡献
回帖0
精华
在线时间 小时
|
一、指南草案的重要意义
2023年9月7日,FDA针对510(k)接连发布3项指南草案,为优化510(k)项目的透明度、可预测性和一致性所迈出的重要一步。
三则指南草案分别为:
1.《选择等同器械以支持上市前通知510(k)提交的最佳实践》
2.《上市前通知510(k)提交的临床数据使用建议》
3.《植入器械510(k)的证据预期》
其中,《选择等同器械以支持上市前通知510(k)提交的最佳实践》这一指南草案尤其受大众关注。如果最终得以确定,该指南草案将极大地改变识别等同器械以做实质等效比较的过程,并将对所有计划提交510(k)或已批准器械的更改产生重大影响。本期,着重解读该指南草案。

二、指南草案提出的最佳实践方法
■ 该指南草案引入4种全新的所谓“最佳实践方法”,用于选择支持510(k)的等同器械。当存在多个“有效等同器械”时,即510(k)提交者可证明实质等效性的合法销售器械(基于预期用途和技术特征),510(k)提交者应使用最佳实践的方法在前述有效等同器械间选择。
根据指南草案,有效的等同器械应采用如下4种最佳的寻找方法:
☑ 已被证明使用公认方法,如:共识标准或FDA指南中的方法;
☑ 考虑设计故障、不良事件的上市后报告,符合或超出安全性和性能的预期;
☑ 不存在“未被缓解的使用或设计相关安全问题”,包括:考虑FDA的安全沟通;
☑ 无设计相关的召回事件影响。
注意:510(k)提交者在提交材料中说明如何选择有效等同器械时,应使用对比表格(识别所有等同器械,并描述上述4方面和实质等同的理由)。
■ 该指南草案已创建相应的新流程,510(k)提交者应通过该流程评估:符合最佳实践的有效等同器械。
■ FDA旨在通过推动创新者依靠更现代的同类器械或客观的性能标准,以促进创新并提高安全性,而考虑最佳实践,将鼓励510(k)计划中更安全、更有效的医疗器械随时间推移而进步。
■ 虽然510(k)程序的现代化可能有益于公共健康,但该指南草案对FDA拟议的新方法的潜在法规和监管影响提出实际挑战。
三、指南草案对法规和监管的挑战
■ 该指南草案若被通过并得以实施,可能与现行法规“关于器械何时不能认定等同的标准”发生重大偏离。
按照现行规定,如果某种器械在FDA倡议下被移除市场或被司法命令认定为品牌错误或掺假,则该器械不能作为等同器械。
尽管CDRH谨慎地将最佳实践描述为“建议”,并非意图对适用的法律和监管标准提出更改,但实践中“建议”会否成为“要求”尚不可知。
■ 指南草案还可能影响实质性、等价性的确定。
根据规定,如果某种器械与等同器械具有相同的预期用途,被证明同样的安全有效,并且不会引发不同的安全性或有效性问题,则该器械为实质等同。
但如果该指南草案被实施,将要求某些510(k)提交者证明:与等同器械对比之下的优越性而非实质等效性。
根据指南草案,如果未根据上述4种最佳方法选择等同器械,510(k)提交者应描述“有效等同器械的所有已知问题,如何通过受试器械得以缓解?”。这实际意味着:510(k)提交者必须证明新器械比等同器械更安全和/或更有效。
而上述方法也被反映于配套的指南草案“510(k)提交中使用临床数据的建议”,该草案表明:当上述新器械自入市以来出现新风险或增加风险时,针对等同器械可能需要提交临床数据,以证明其实质等效性。
→ 欢迎点击上方粉我~关注【久顺医械技术服务】医械知识不迷路! |
|

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 






 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 发表于 2025-1-25 06:13
发表于 2025-1-25 06:13






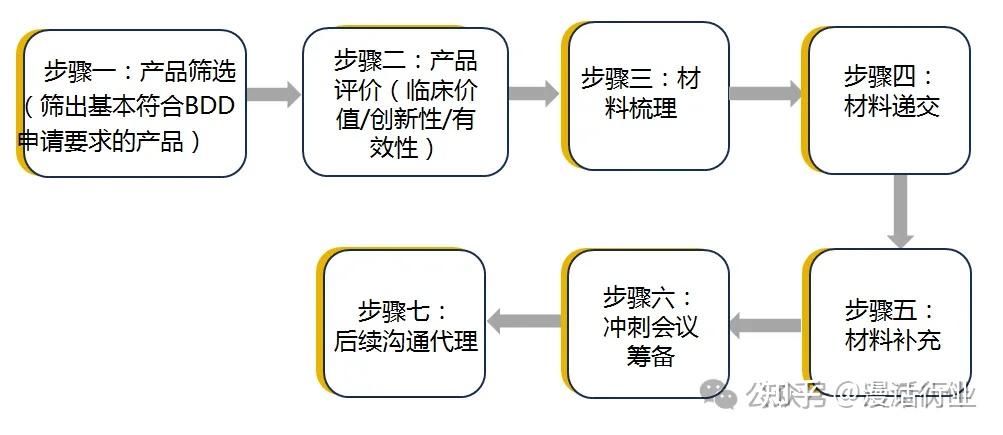

 发表于 2025-1-25 06:16
发表于 2025-1-25 06:16



