金桔
金币
威望
贡献
回帖0
精华
在线时间 小时
|
基因编辑意味着能够人为改造遗传物质,特别是对特定片段的敲除、加入等,与基因工程有一些类似之处。当前,最抢眼的基因编辑技术就是CRISPR/Cas9了,CRISPR就是clustered regularly interspaced short palindromic repeats,即成簇规律性间隔重复序列。
CRISPR是由短的(大概20~50碱基对(bp)左右)DNA重复序列形成的断续结构组成的,这部分存在回文(回文指的就是12321,2378732,AGTTCCTTTCCTTGA这样的结构,进化上形成高度回文的序列,可作为一种保护基因免遭遗传漂变和突变轰炸的策略,在人类的Y染色体上就发现了9个大规模(可长达百万pb)回文结构)排布,重复序列之间被25~70碱基对左右的非重复序列隔开。重复的部分和间隔的部分分别称为重复序列(repeats)和间隔序列(spacer),此外参与CRISPR构成的还有一个具有类似启动子的前导(leader)序列。CRISPR-Cas系统存在于至少40%的细菌和90%的古生菌中,是细菌用以记忆、识别和抵抗噬菌体的系统。而正是它能够修改遗传物质,进而将病毒基因从自己的基因组内踢出的能力引起了注意。Cas就是CRISPR-associated sequence即CRISPR关联序列,它是由好几个与CRISPR相关的基因组成的序列。
不过,CRISPR/Cas9并不是唯一的基因编辑技术。在此之前还出现过锌指核酸酶(zinc-finger nucleases)和转录激活因子样效应核酸酶(transcription activator-like effector nucleases)等,这两种基因编辑技术,题主可自行了解,这里就只介绍CRISPR/Cas(9)系统(技术)了。
CRISPR/Cas系统最初是日本人在研究一种E.coli蛋白的时候发现的,2002年才被Jansen命名。它有I,II,III三种类型,类型I广泛分布,在Cas3(一种核酸酶)的参与下降解外源DNA;类型II只在细菌中发现,由Cas9(也是核酸酶)参与,类型III还分A,B两种亚型,比较复杂,它们则似乎是古生菌特有的。在它们当中,Cas9最容易技术化(因为它单独行动,无需跟其他核酸酶合作),所以就有了CRISPR/Cas9技术(当然,还是经过了改造以后才得来的)。
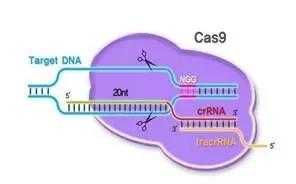
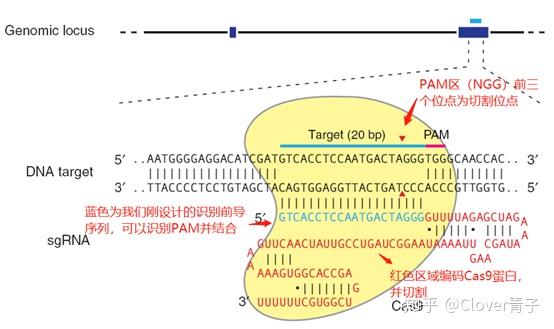
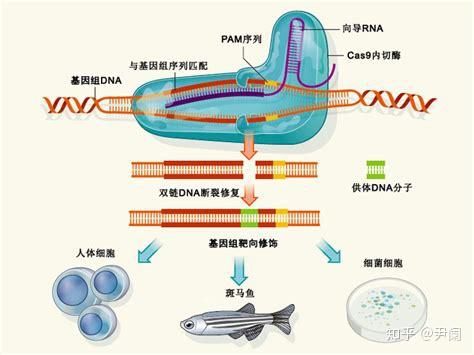
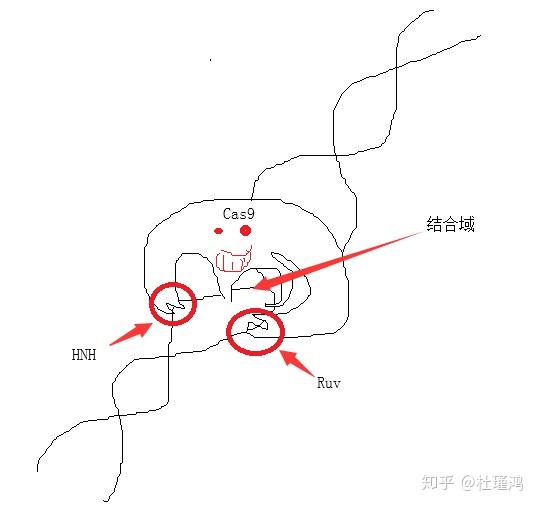
细菌在识别外源序列(如何识别,细菌当然自有策略)以后,Cas蛋白就会出动。Cas9蛋白质的身上有两个部分(Ruv和HNH),相当于“剪刀”,被用来切割DNA(当然是先与之结合,把自己固定在目标DNA上,再大刀阔斧地进行切割),这两个部分分别负责一条链(这种快速而简单的切割方式,正是它受到青睐的原因),即便因为这两个部分发生突变失去切割能力(这样的Cas9就叫做dead Cas9简称dCas9),Cas9依然可以在sgRNA介导之下,通过与靶基因结合,阻断它的转录或者干脆使之无法转录。如果觉得不够直观,可以见下图。
其他Cas蛋白复合物会负责割取目标DNA的一些小段,作为间隔序列插入到自己的CRISPR部分,就实现了对外源DNA的记忆,但又不至于受到损害。如果同类噬菌体再度入侵,相应的CRISPR序列迅速转录生成pre-crRNA(即crRNA前体),Cas复合体与另外一种RNA(tracrRNA)作用剪切这个前体,生成crRNA,(是不是与hnRNA最终被剪切成mRNA有异曲同工之妙?)crRNA与前两者形成复合体,就可以识别和剪切与crRNA互补的位点。(在细胞中,其实一部分机制与此类似的阻碍(调节)蛋白质合成的DISC即RNA诱导沉默复合体,也能帮助免疫系统相对脆弱的昆虫和植物抵抗病毒。)
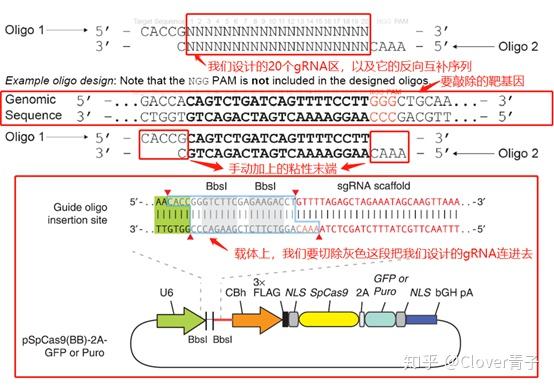
那么实验人员是怎么改造Cas9的呢?其实介绍了细菌体内CRISPR/Cas9的功能,它的技术改造就很好说了。上面提到crRNA在形成后与tracrRNA与Cas9形成三聚复合体。但是事先,tracrRNA和crRNA会结合成二聚体。然而在细胞里,需要稍作改变。我们必须设计一种sgRNA代替这种二聚体,与Cas9形成另外一种二聚体。这种sgRNA是从宿主细胞的意义上来说的,以为它必须与靶基因匹配,才能使Cas9顺利行使切割功能。然后,还需要一个用来定位的装置,那就是核定位信号。无需准备修复策略,因为细胞本身自有办法。(详细的技术机制,我有时间再写。)
但是由于这是一项新兴技术,所以它还存在诸多缺陷,包括无法避免因种间差异而引发的效率挫伤,脱靶之类,所以还需更多改进。
当然,在这篇回答里我可能讲得还不是很清楚。你可以看一看我写的一篇文章:
杜瑾鸿:核酸的剪刀——CRISPR/Cas
参看文献:
Ishino, Y., Shinagawa, H., Makino, K., et al. (1987) Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. Journal of Bacteriology,169, 5429-5433.
Jansen, R., Embden, J.D., Gaastra, W., et al. (2002). Identification of genes that are associated with DNA repeats in prokaryotes. Molecular Microbiology, 43, 1565-1575
Wei, C.X., Liu, J.Y., Yu, Z.S., et al. (2013) TALEN or Cas9-rapid, efficient and specific choices for genome modifica[1]tions. Journal of Genetics and Genomics, 40, 281-289 |
|

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 






 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 发表于 2025-1-24 23:06
发表于 2025-1-24 23:06