金桔
金币
威望
贡献
回帖0
精华
在线时间 小时
|
RNA/DNA提取过程中的注意事项
1.避免污染
为避免RNase/DNase等污染,可以在操作前仔细清洁实验平台、试剂盒、器皿和工具等,并使用经RNase/DNase清洗的试剂和器皿。此外,应该尽可能快地进行样品提取,以减少RNA/DNA降解和污染的可能性。
2.明确目的和选择合适的方法
RNA和DNA在分子结构和物理化学特性上有所不同,并且不同的样本类型也有不同的组织结构和特征,因此需要根据实验目的和样本类型选择合适的RNA/DNA提取方法。例如,在提取RNA时,需要更加注意RNase的污染问题;而在提取DNA时,则需要更加注意DNA的不断降解问题。
3.优化样品破碎步骤
样品破碎对RNA/DNA提取至关重要。不同样品类型需要不同的破碎方法,如高压均质机、超声波等。在破碎过程中,需要注意破碎时间和功率控制,以避免过度破碎导致RNA/DNA的降解。
细胞破碎方法:超声波法、高压均质法、玻璃珠法等都是常用的细胞破碎方法。
4.RNA/DNA的纯化和浓缩
在提取和纯化RNA/DNA过程中,需要去除可能存在的污染物,如蛋白质、盐等,并尽可能地减少DNA和RNA的降解。此外,在进行RNA/DNA浓缩时,应根据实验要求选择合适的方法,如酚/氯仿法或柱层析法等。
5.对提取的RNA/DNA进行检测和评估
在提取RNA/DNA之后,需要对其进行确定性和质量评估。可以使用分光光度计和凝胶电泳等技术来测定RNA/DNA的浓度和质量,并评估是否存在RNase/DNase污染等问题。
6.RNase/DNase污染:
RNase或DNase的污染很容易导致RNA/DNA降解,因此需要使用经RNase/DNase清洗的器皿和试剂,并在操作中避免污染。
7.温度控制:
RNA/DNA在高温下易于分解,因此在提取RNA/DNA时要控制好温度。在冷冻样品之前,通常建议在低温环境中培养细胞或收集组织,以最大程度地保护RNA/DNA。
8.RNA/DNA保存:
RNA/DNA应该储存在-80℃冰箱中,以避免降解和污染。在长期储存之前,建议进行浓缩,并将RNA/DNA用RNase-free水稀释至合适的浓度。
9.质检:
在提取RNA/DNA之后,需要对其进行确定性和质量评估。可以使用分光光度计来测定RNA/DNA的浓度和质量,或者使用凝胶电泳来确认RNA/DNA的大小和纯度.
RNA/DNA提取的常见问题及解决方法:
1.RNA/DNA浓度低:
如果提取得到的RNA/DNA浓度较低,则可以通过浓缩RNA/DNA溶液或在提取过程中增加样品量来提高RNA/DNA的浓度。
2.RNA/DNA质量不佳:
如果RNA/DNA分离后质量不佳,则可能是由于RNase/DNase污染、过度破碎、过度处理等原因导致的。需要仔细检查每个步骤,并采取相应的措施来避免这些问题。
3.提取RNA或DNA有选择性:
有时,RNA或DNA的提取会出现选择性问题,即只能提取其中一种。这通常是由于使用的试剂或条件不适合同时提取RNA和DNA所导致的。可以尝试更换试剂或优化条件,以获得更好的RNA/DNA提取结果。
4.样品残留:
在提取RNA/DNA时,倒掉废液后可能会有样品残留在管子或器皿中。这可能会导致RNA/DNA的污染和降解,因此需要特别小心地清洁所有器皿和工具,确保没有任何残留物。
总之,在进行RNA/DNA提取时需要注意许多方面,并且可能会遇到一些问题。如果出现问题,需要仔细检查每个步骤并逐步解决问题,以确保最终得到高质量的RNA/DNA样本。
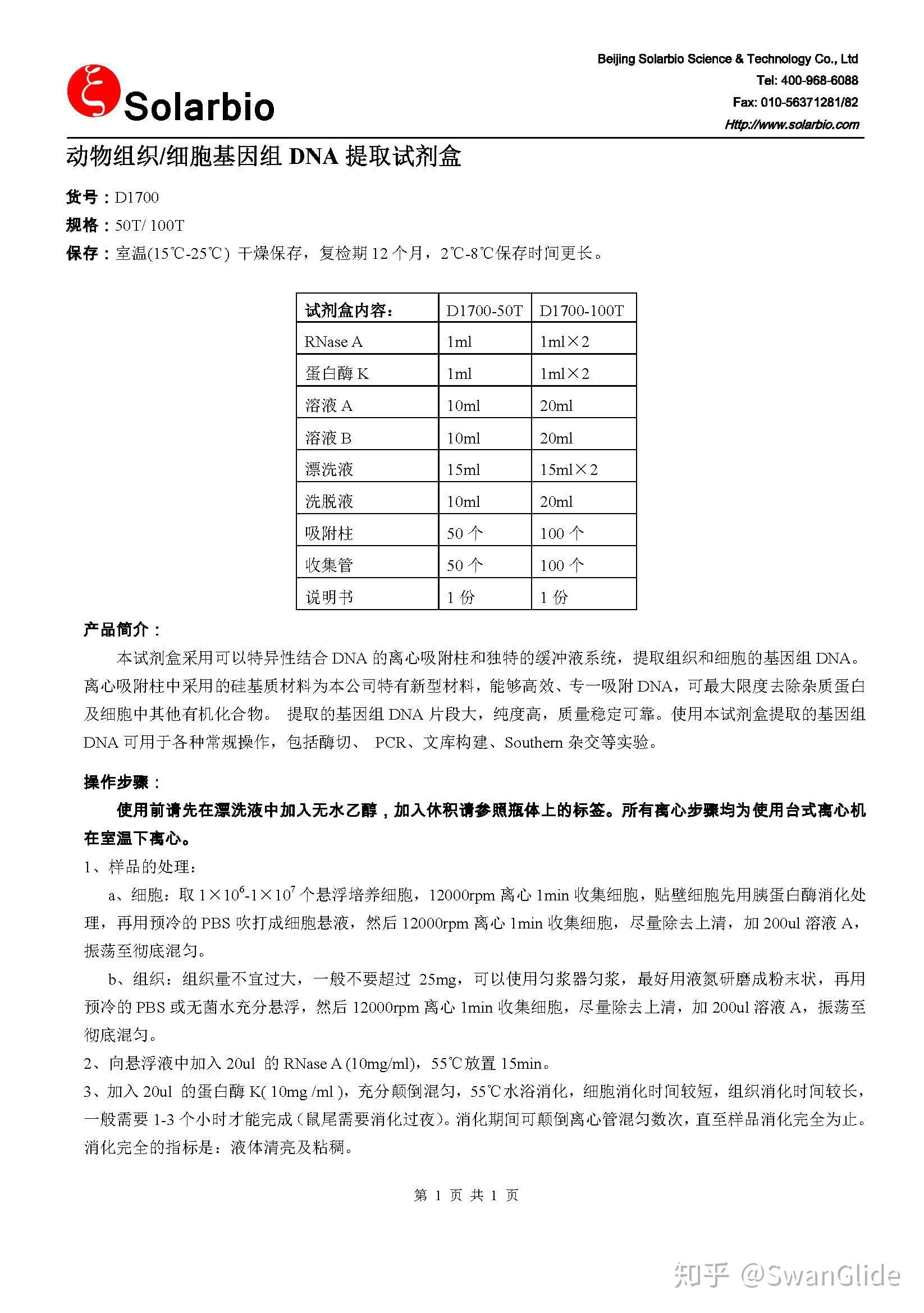
动物组织样本DNA提取步骤
1.动物组织DNA提取准备
① 材料:动物组织
② 试剂:TIANGEN血液/细胞/组织基因组DNA提取试剂盒,无水乙醇等
③ 设备:离心机,微量移液器等
2.动物组织DNA提取步骤
① 处理材料:取动物组织10mg,尽量切碎,加200μl缓冲液GA,振荡至彻底悬浮。
② 加入20μl蛋白酶K溶液,混匀后56℃水浴,直至组织溶解(不同组织裂解时间不同,通常需1-3小时即可完成)。简短离心以去除管盖内壁的水珠,再进行下一步骤。
③ 加入200μl缓冲液GB,充分颠倒混匀,70℃放置10分钟,溶液应变清亮,简短离心以去除管盖内壁水珠。(溶解DNA)
注意:加入缓冲液GB时可能会产生白色沉淀,一般70℃放置时会消失,不会影响后续实验。如溶液未变清亮,说明细胞裂解不彻底,可能导致提取DNA量少和提取出的DNA不纯。
④ 加入200μl无水乙醇,充分振荡混匀15s,此时可能出现絮状沉淀,简短离心以去除管盖内壁的水珠。(沉淀DNA)
⑤ 将上一步所得溶液和絮状沉淀都加入一个吸附柱CB3中,吸附柱放入收集管中,12,000rpm(~13,400×g)离心30s,倒掉废液,将吸附柱CB3放回收集管中。(吸附沉淀)
⑥ 向吸附柱CB3中加入500μl缓冲液GD(使用前先检查是否已加入无水乙醇),12,000rpm(~13,400×g)离心30s,倒掉废液,将吸附柱CB3放回收集管中。
⑦ 向吸附柱CB3中加入700μl漂洗液PW(使用前先检查是否已加入无水乙醇),12,000rpm(~13,400×g)离心30s,倒掉废液,将吸附柱CB3放回收集管中。
⑧ 向吸附柱CB3中加入500μl漂洗液PW,12,000rpm(~13,400×g)离心30s,倒掉废液。(6、7、8为漂洗)
⑨ 将吸附柱CB3放回收集管中,12,000rpm(~13,400×g)离心2min,倒掉废液。将吸附柱CB3置于室温放置数分钟,以彻底晾干吸附材料中残余的漂洗液。(注意:这一步的目的是将吸附柱中残余的漂洗液去除,漂洗液中乙醇的残留会影响后续的PCR等实验)
⑩ 将吸附柱CB3转入一个干净的离心管中,向吸附膜的中间部位悬空滴加50-200μl洗脱缓冲液TE,室温放置2-5min,12,000rpm(~13,400×g)离心2min,将溶液收集到离心管中。(溶解洗脱DNA)(注意:采用硅基质膜吸附的DNA,可将DNA在低盐高pH值条件下洗脱下来。pH值在7.0-8.5之间洗脱效率较高,pH值低于7.0则洗脱效率很低)
洗脱缓冲液体积不应该少于50μl,体积过小影响回收效率。为增加基因组DNA的得率,可将离心得到的溶液再加入吸附柱CB3中,室温放置2min,12,000rpm(~13,400×g)离心2min。DNA产物应保存在-20℃,以防止DNA降解。
植物组织样本DNA提取步骤
1.植物组织DNA提取仪器与设计准备
实验仪器:高速离心机;烘箱;冰箱;水浴锅;高压灭菌锅;Nanodrop。
实验试剂:玻璃珠,十二烷基磺酸钠(SDS);三羟甲基氨基甲烷(Tris);乙二胺四乙酸
(EDTA);氯化钠;苯酚;氯仿;无水乙醇等。
2.植物组织DNA提取步骤
植物组织的DNA提取通常采用机械研磨的方法破碎植物的组织和细胞,由于植物细胞匀浆含有多种酶类(尤其是氧化酶类)对DNA的抽提产生不利的影响,在抽提缓冲液中需加入抗氧化剂或强还原剂(如巯基乙醇)以降低这些酶类的活性。在液氮中研磨,材料易于破碎,并减少研磨过程中各种酶类的作用。
详细操作步骤如下:
① SDS提取缓冲液在65℃水浴中预热。
② 将叶片置于1.5ml离心管中,液氮速冻,组织研磨器打样。
③ 加入700ml的SDS提取缓冲液,涡旋摇匀。
④ 置于65℃的水浴中,每隔10min轻轻摇动,30min后取出。
⑤ 加入200mlKAc溶液,摇匀,放入-20℃冰箱30min。
⑥ 10000rpm离心5min,上清移至新离心管中,12000rpm离心5min。
⑦ 上清移至新离心管中,加入700ml异丙醇,-20℃冰箱30min。
⑧ 10000rpm离心5min,弃上清,加入500ml70%乙醇漂洗。
⑨ 弃液体,晾干。
⑩ DNA纯度与浓度测定(使用nanodrop)。
DNA纯度:DNA的OD260/OD280一般为1.8左右。
OD260/OD280»1.8:说明较纯;
OD260/OD280>1.8:说明可能有RNA污染;
OD260/OD280<1.8:说明可能有蛋白质污染。
3.植物组织DNA提取注意
① 叶片磨得越细越好。
② 注意移液器的正确使用。
③ 由于植物细胞中含有大量的DNA酶,因此,除在抽提液中加入EDTA抑制酶的活性外,第一步的操作应迅速,以免组织解冻,导致细胞裂解,释放出DNA酶,使DNA降解。
4.RNA提取注意事项
① 组织样本要尽可能的新鲜,避免反复冻融。
② 提取时组织要充分研磨,组织量不宜过少,更不宜过多。
③ 加入裂解液后应给予充分的孵育时间,使样本充分裂解。
④ 在用Trizol法提取时,分层后吸取上清的原则是“宁愿少吸,不能多吸”,千万不能提取到中间层,否则会导致严重的基因组DNA污染。
⑤ 洗涤时洗涤液应充分浸润到管壁的四周,确保洗涤彻底。
⑥ 柱式提取法,洗涤完后除了对柱子进行空离后,还应当将吸附柱置于超净台内吹风5~10 min,使有机溶剂充分挥发干。
⑦ 柱式法最后洗脱时候,当加入DEPC水后还应孵育3~5 min,或者提前将DEPC水加热至60℃,可提高洗脱得率。传统的Trizol裂解异丙醇沉淀法最后RNA是用DEPC水溶解沉淀,则应当给予适当溶解时间,并用枪头不断吹吸离心管底部。
其他原因及解决办法
1.获得RNA效率低有一下原因
① 样品裂解或匀浆处理不彻底
② 最后得到地RNA沉淀未完全溶解
2.A260/A280<1.65原因
① 检测吸光度时,RNA样品不是溶于TE,而是溶于水。低离子浓度和低PH条件下,A280值会较高
② 样品匀浆时加地试剂量太少
③ 匀浆后样品未在室温放置2分钟
④ 水相中混有有机相
⑤ 最后得到地RNA沉淀未完全溶解
3.RNA降解原因
① 组织取出后没有马上处理或冷冻
② 样品或提取地RNA沉淀保存于-5--20度,未在-60--70度保存
③ 细胞在胰酶处理时被破坏
④ 溶液或离心管未经RBase去处理
4.预防RNase污染,应注意以下几个方面
① 经常更换新手套。因为皮肤经常带有细菌,可能导致RNase污染。
② 使用无RNase的塑料制品和枪头,避免交叉污染。
③ RNA在Trizol试剂中不会被RNase降解。但提取后继续处理过程中应使用不含RNase的塑料和玻璃器皿。玻璃器皿可在150 ℃烘烤4h,塑料器皿可在0.5M NaOH中浸泡10min,然后用水彻底清洗,再灭菌,即可去除RNase。
④ 配制溶液应使用无RNase的水(将水加入到干净的玻璃瓶中,加入DEPC至中浓度0.1%(v/v),放置过夜,高压灭菌。注:DEPC有致癌之嫌,需小心操作)。 |
|

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 





 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 雷达卡
雷达卡 发表于 2025-1-24 11:42
发表于 2025-1-24 11:42


 提升卡
提升卡 发表于 2025-1-24 11:42
发表于 2025-1-24 11:42