用户名
UID
Email
密码
记住
立即注册
找回密码
只需一步,快速开始
微信扫一扫,快速登录
开启辅助访问
收藏本站
快捷导航
门户
Portal
社区
BBS
资讯
会议
市场
产品
问答
数据
专题
帮助
签到
每日签到
企业联盟
人才基地
独立实验室
产业园区
投资机构
检验科
招标动态
供给发布
同行交流
悬赏任务
共享资源
VIP资源
百科词条
互动话题
导读
动态
广播
淘贴
法规政策
市场营销
创业投资
会议信息
企业新闻
新品介绍
体系交流
注册交流
临床交流
同行交流
技术杂谈
检验杂谈
今日桔说
共享资源
VIP专区
企业联盟
投资机构
产业园区
业务合作
投稿通道
升级会员
联系我们
搜索
搜索
本版
文章
帖子
用户
小桔灯网
»
社区
›
C、IVD技术区
›
Western Blotting技术
›
关于western blot,你不知道的信息
图文播报
2026庆【网站十三周
2025庆【网站十二周
2024庆中秋、迎国庆
2024庆【网站十一周
2023庆【网站十周年
2022庆【网站九周年
返回列表
查看:
7836
|
回复:
0
[分享]
关于western blot,你不知道的信息
[复制链接]
幸福侠侣
幸福侠侣
当前离线
金桔
金币
威望
贡献
回帖
0
精华
在线时间
小时
雷达卡
发表于 2025-1-4 14:26
|
显示全部楼层
|
阅读模式
登陆有奖并可浏览互动!
您需要
登录
才可以下载或查看,没有账号?
立即注册
×
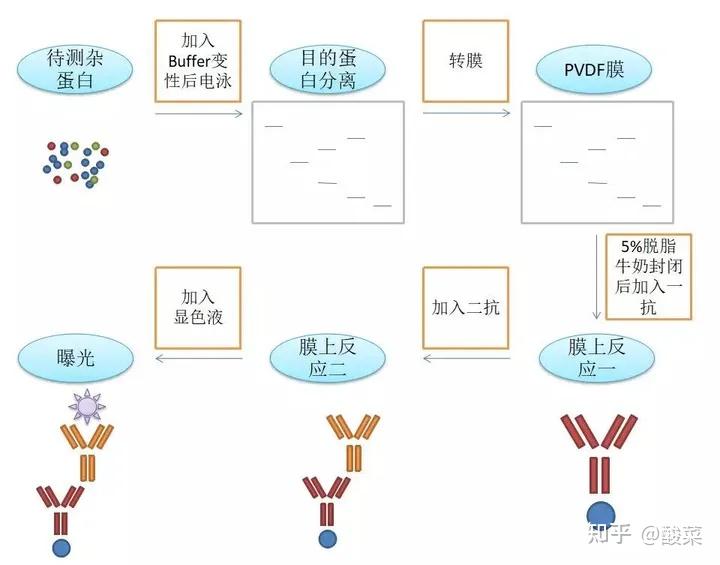
Western blot,中文为蛋白质免疫印迹试验,简单来说是
抗原抗体
特异性结合。
电泳分离后的蛋白转移到固相载体PVDF膜上,以共价键的形式吸附蛋白质,并作为抗原结合
特异性抗体
。二抗作为显色标记,经过底物显色反应组织和细胞中蛋白的表达情况。
蛋白提取
组织蛋白提取
相对于细胞而言,组织蛋白的提取对技术和外界环境要求会更高一些。因为我们养的细胞很多是根据具体实验选择的特定细胞,而组织大多是为了丰富体内实验的各种蛋白的混合体,目的蛋白的含量并不一定充分,而且其中还包含着丰富的蛋白酶、血液或者脂肪等干扰因素。 所以个人认为,提取组织蛋白一定要做预实验,这是很有必要的。可以摸清楚每次应该取多少重量的组织,什么样的离心条件可以去除血液和脂肪的干扰。
比如像心脏、脾这类血液较多的组织,剪成米粒大小的小块儿后,加入适量PBS(PBS:PMSF=100:1,PMSF为
苯甲基磺酰氟
,蛋白酶抑制剂,30min内会降解一半,因此每次提取蛋白要加新鲜的PMSF),7500g离心力,离心5分钟,去上清。
如果感觉血液还是很多,可以重复上述步骤数次。
蛋白提取的全程要在低温环境下进行,以防止蛋白降解。
30mg的组织对应500ul的RIPA裂解液(RIPA全称为Radio Immunoprecipitation Assay,RIPA:PMSF=100:1),置于冰上,使用研磨棒或电动组织研磨器充分研磨30分钟,再置于冰上静止30分钟。
在12000rpm转速下,4度离心15分钟,取上清,测蛋白浓度。
2.细胞蛋白提取
细胞蛋白的提取相对来说耗时会少很多,只是在贴壁细胞和悬浮细胞的处理方式上略有不同。 对于贴壁细胞而言,去除培养基后,用无菌预冷的PBS清洗3次,每次3分钟。对于像6cm的培养皿,我习惯每次加100ul的RIPA裂解液(RIPA:PMSF=100:1),转动培养皿,让裂解液接触到整个皿面。之后用细胞刮刀将皿里的细胞刮刀一侧,冰上静置15分钟后,在12000rpm转速下,4度离心15分钟,取上清,测蛋白浓度。 对于悬浮细胞而言,吸出培养皿中的培养基,按照传代时离心力大小和时间进行离心。离心结束后,小心的倒掉上清,加入100ul的RIPA裂解液(RIPA:PMSF=100:1),冰上静置15分钟后,在12000rpm转速下,4度离心15分钟,取上清,测蛋白浓度。
3.测定蛋白浓度
蛋白浓度的测定一般选用BCA工作液(bicinchoninic acid),BCA工作液作为一种稳定的水溶性复合物,可以与二价铜离子结合,产生紫色复合物。562nm波长条件下测定吸光度,制作标准曲线,吸光度与蛋白浓度成正比。
4.蛋白变性
根据蛋白的体积,计算加入多少体积的1*loading buffer,煮沸5分钟即可。Loading buffer中主要含有溴酚蓝、二甲苯氰和甘油。前两者起到指示作用,甘油起到沉降蛋白的作用,以防加样时蛋白飘出。
电泳
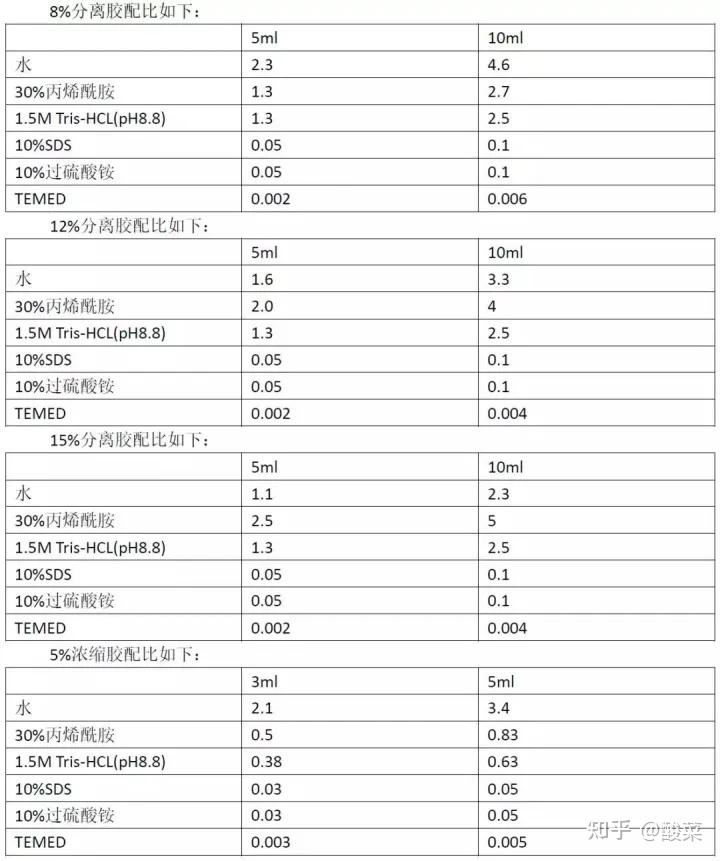
1.制胶
根据目的蛋白的分子量大小选择不同浓度的分离胶,8%、12%和15%三种浓度的分离胶就完全可以搞定日常蛋白的测定。一般测定100kDa分子量以上的蛋白可选择8%浓度的分离胶;测定20kDa分子量以下的蛋白可选择15%浓度的分离胶,中间的分子量可选用12%浓度的分离胶。 有的时候蛋白跑不出来,其实未必是分离胶浓度选的不好,也有可能是marker的问题。marker起到的是蛋白分子量指示剂作用,但是要根据蛋白分子量选择合适的
marker
。一般的marker分子量范围在10-130kDa之间,都可以满足正常蛋白的需要。 但有些分子量范围较广的marker,比如说6-250kDa,虽然说它范围很广,可中间的蛋白分子量并没有区分的很细,只适用于大分子或小分子蛋白,而对于30-70 kDa的蛋白并不是很适用。
电泳条件
个人使用的条件一直是80V,300mA跑浓缩胶,120V,300mA跑分离胶,恒压电泳。
当然了,这个电泳条件不是固定的,也有人选择恒流跑电泳。个人认为恒流和恒压对电泳的效果影响没有什么区别。因为配胶的成分中SDS起到的作用是破坏蛋白质分子和其它物质分子之间的
非共价键
。
解聚后的蛋白质分子与SDS形成的复合物带有负电荷,远远超过蛋白质本身的电荷量,所以就不存在不同蛋白质分子之间的电荷量不同造成的影响了,恒压或恒流都只是起到推动作用。
至于浓缩胶和分离胶的条件不同,我想这一点大家应该都很清楚了。浓缩胶中的孔隙较大,起到的作用是为了让蛋白质混合物在进入分离胶之前都到达同一起跑线,以便在分离胶中更好的分开,因此一定要充分跑完浓缩胶之后再调电压。
转膜
转膜分为
湿转
和干转。湿转就是把三明治结构放在转膜液中进行;干转,其实应该是半干转,虽然不放在转膜液中进行,但是滤纸也要事先泡过转膜液。有人说,湿转主要用大分子蛋白质转膜,半干转主要用于小分子蛋白质转膜,但是个人认为湿转就完全可以搞定大小所有分子量的蛋白,而且从转膜稳定率来说要好于半干转。
所以个人习惯于用湿转。
一般情况下的转膜条件是220V,300mA,转膜时间是1小时20分钟。
对于10-180kDa分子量的蛋白,这个条件是完全可以的。虽然网上有很多人说小分子量的蛋白需要缩短转膜时间,或者改变电压电流,但是本人试过用这个条件转8kDa的蛋白质,一样没有问题。相反的,我缩短了转膜时间,或者改成了100V,300mA的转膜条件,转膜的效果反而不好。
所以个人认为,
转膜效果的好坏除了要考虑机器本身设定的条件外,还需要考虑一下其他因素。
有很多新手用完转膜液忘记放回4度冰箱,时间久了,
转膜液
的离子成分就会发生改变,影响转膜效果;或者是滤纸使用太久了,也会影响转膜效果;又或者是要研究的蛋白自身性质决定的。Western blot并不适用于所有的蛋白,对于检测一些分泌型的蛋白,ELISA会更适合,所以也并不是说蛋白实验一定要用Western blot。
发光
选用ECL发光液发光。ECL发光液分为A液和B液,按照1:1的条件混合配制。
洗膜结束后,倒出多余的TBST,但是也要在洗膜的塑料盘中留有一些,防止
PVDF膜
干掉。基本上1ml的工作液可以覆盖10cm3,发光的效果与发光液的新鲜程度有关,所以要现用现配。
ECL的发光原理是:发光剂中的
鲁米诺
被过氧化氢和HRP氧化,产生荧光,整个发光过程被增强剂增强了发光效果,可以将灵敏度提高到1000-100000倍,但是也会遇到猝灭的情况,除了成像机器老化的原因外,也与发光液的新鲜程度和
一抗二抗
的浓度有关。
质量好的发光液配合着合适的一抗二抗浓度,才能快速完美的发光。
小于1分钟的发光时间固然是我们所向往的,但是碰上信号低的蛋白,我们也要想尽办法让它们显示出结果。对于信号弱的蛋白条带,我们可以手动调整发光时间少一些,比如说10sec,快速曝光比延长曝光时间的显影效果更好。
用各种软件处理分析蛋白条带
1.Photoshop CS6
PS软件在后期投稿整理图片方面会有很大作用,这里我想说的是利用PS改变条带的对比度,减少后续利用Image j对蛋白灰度值进行测定时的干扰。
正常机器发光之后的图片,背景都会泛蓝,我们可以选择PS软件中,图像-调整-去色,把条带背景统一调成灰色后再测灰度值。
2.ImageJ
利用Image j测量蛋白灰度值的方法,我想大家都很熟悉了,我就不详细的列出每一步了。
这里想提到一点,BCA测出的蛋白浓度有时会存在一定的误差。我们在测蛋白标准曲线时,得出的R值肯定是9越多越准确,但是实验过程中,我们往往只有一或两个9,导致内参发出来的效果也是各组略有差异。为了追求完美,我们可以通过内参的灰度值进行作比,以一个组的蛋白浓度为定量,适当调整其他各组
上样量
即可。
如果初入实验室时,能够接手师兄师姐的实验,顺带用用师兄师姐传下来的protocol就很幸运了。那要是很惨的被放养,自己摸索protocol,可就要走一步看一步了,毕竟网上五花八门的protocol,信息参差不齐,步步是坑,这要是没有一份靠谱protocol傍身,迟早要脚下一黑的。
为了帮助科研粉丝们少走弯路,解螺旋整理出
实验 Protocol 资源包!标书、毕业论文都能用上!
包括
不同研究水平实验技术Protocol,常见细胞动物实验技术总结,不同实验方法全流程,WB实验流程、注意事项、数据处理及写作,IHC实验流程、操作技巧、注意事项、图像分析等
,全都是经过前辈们无数次验证过的,绝对靠谱!
今天给大家带来
实验protocol资源包,可以直接拉到文末领取。
部分截图:
1
不同研究水平实验技术protocol
针对不同研究水平的同学,我们提供不同的实验技术Protocol,帮助科研er解决实验屡战屡败难题,拓展研究思维并发展未来研究新方向。
入门↓↓↓
初级↓↓↓
中阶↓↓↓
打包送大家一个医学SCI从入门到精通的免费训练营名额。如果你也想轻松搞定实验,高效发SCI,赶紧点击免费领取吧!
原文地址:https://zhuanlan.zhihu.com/p/605732846
回复
使用道具
举报
提升卡
返回列表
发表回复
高级模式
B
Color
Image
Link
Quote
Code
Smilies
您需要登录后才可以回帖
登录
|
立即注册
本版积分规则
发表回复
回帖后跳转到最后一页
关闭
官方推荐
/3
AI助手<小桔子>来了!
欢迎来交流,可以回答IVD行业各类问题!
查看 »
IVD业界薪资调查(月薪/税前)
长期活动,投票后可见结果!看看咱们这个行业个人的前景如何。请热爱行业的桔友们积极参与!
查看 »
小桔灯网视频号开通了!
扫描二维码,关注视频号!
查看 »
返回顶部
快速回复
返回列表
客服中心
搜索
洽谈合作
关注微信
微信扫一扫关注本站公众号
个人中心
个人中心
登录或注册
业务合作
-
投稿通道
-
友链申请
-
手机版
-
联系我们
-
免责声明
-
返回首页
Copyright © 2008-2024
小桔灯网
(https://www.iivd.net) 版权所有 All Rights Reserved.
免责声明: 本网不承担任何由内容提供商提供的信息所引起的争议和法律责任。
Powered by
Discuz!
X3.5 技术支持:
宇翼科技
浙ICP备18026348号-2
浙公网安备33010802005999号
快速回复
返回顶部
返回列表

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 





 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 雷达卡
雷达卡 发表于 2025-1-4 14:26
发表于 2025-1-4 14:26








 提升卡
提升卡


