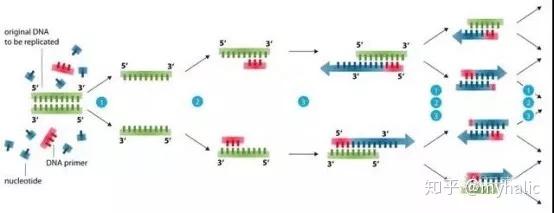
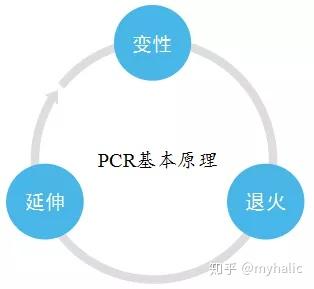
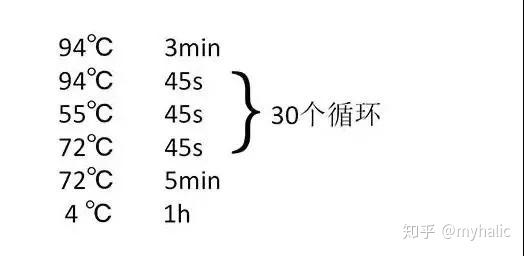
1模板 DNA 的变性:模板 DNA 经加热至 94℃ 左右一定时间后,使模板 DNA 双链或经 PCR 扩增形成的双链 DNA 解离,使之成为单链,以便它与引物结合,为下轮反应做准备;
2模板 DNA 与引物的退火 (复性):模板 DNA 经加热变性成单链后,温度降至 55℃ 左右,引物与模板 DNA 单链的互补序列配对结合;
3引物的延伸:DNA 模板--引物结合物在 Taq 酶的作用下,以 dNTP 为反应原料,靶序列为模板,按碱基配对与半保留复制原理,合成一条新的与模板 DNA 链互补的半保留复制链。
重复循环变性--退火--延伸三过程,就可获得更多的「半保留复制链」,而且这种新链又可成为下次循环的模板。每完成一个循环需 2~4 分钟, 2~3 小时就能将待扩目的基因扩增放大几百万倍。

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 






 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 发表于 2025-1-3 10:27
发表于 2025-1-3 10:27