用户名
UID
Email
密码
记住
立即注册
找回密码
只需一步,快速开始
微信扫一扫,快速登录
开启辅助访问
快捷导航
门户
Portal
社区
BBS
资讯
会议
市场
产品
问答
数据
专题
帮助
签到
每日签到
企业联盟
人才基地
独立实验室
产业园区
投资机构
检验科
招标动态
供给发布
同行交流
悬赏任务
共享资源
VIP资源
百科词条
互动话题
导读
动态
广播
淘贴
法规政策
市场营销
创业投资
会议信息
企业新闻
新品介绍
体系交流
注册交流
临床交流
同行交流
技术杂谈
检验杂谈
今日桔说
共享资源
VIP专区
企业联盟
投资机构
产业园区
业务合作
投稿通道
升级会员
联系我们
搜索
搜索
本版
文章
帖子
用户
小桔灯网
»
社区
›
C、IVD技术区
›
基因编辑技术
›
关于基因编辑,目前有哪些发展进度?
图文播报
2026庆【网站十三周
2025庆【网站十二周
2024庆中秋、迎国庆
2024庆【网站十一周
2023庆【网站十周年
2022庆【网站九周年
返回列表
查看:
10437
|
回复:
1
[分享]
关于基因编辑,目前有哪些发展进度?
[复制链接]
空白派
空白派
当前离线
金桔
金币
威望
贡献
回帖
0
精华
在线时间
小时
发表于 2024-12-25 14:34
|
显示全部楼层
|
阅读模式
登陆有奖并可浏览互动!
您需要
登录
才可以下载或查看,没有账号?
立即注册
×
想了解一下基因编辑有那些进展,我们目前在人类基因编辑上可以实现哪些突破?
原文地址:https://www.zhihu.com/question/7192478959
回复
举报
长长的路
长长的路
当前离线
金桔
金币
威望
贡献
回帖
0
精华
在线时间
小时
发表于 2024-12-25 14:35
|
显示全部楼层
这个问题问的过于宏观啊,我推荐你找个综述看。
比如这篇,苏黎世大学的研究人员在
Cell
发的综述,Past, present, and future of CRISPR genome editing technologies(CRISPR基因组编辑技术的过去、现在和未来)。
全文很长有一万多字,可以系统性的了解基因编辑的历史现状和未来发展趋势,对于你感兴趣的目前的发展进度,可以搜索“基础研究和人类医学中的应用”这部分来看,有精力的话可以分多次读完这篇文章,还是很有价值的。
CRISPR基因组编辑技术的过去、现在和未来
摘要
基因组编辑是生命科学和人类医学中的一项变革性技术,为解析复杂的生物学过程和治疗许多遗传疾病的根本原因提供了前所未有的机会。基于CRISPR的技术以其显著的效率和易于编程的特性,站在这场革命的前沿。本文综述了CRISPR基因编辑技术在研究和治疗中的当前状况,分析了限制其应用的局限性,以及近年来为解决这些问题而开发的技术创新。此外,文章总结了基因编辑在人类健康和治疗领域的应用现状,并展望了可能在未来塑造基因编辑技术及其应用的潜在发展方向。
引言
基因组编辑,即对生物体遗传物质的精确和靶向修改,代表了分子生物学领域最重要的进步之一。它的应用范围非常广泛,从揭示基本的生物学过程到推动医学、农业和生物技术的发展。2023年末,第一个基于CRISPR的基因编辑疗法获批,这标志着CRISPR基因组编辑技术进入了一个新的时代。本文旨在全面回顾CRISPR基因组编辑的研究领域,重点分析其当前的发展状况、未来的可能进展以及实现其在人类医学应用中需要克服的障碍。
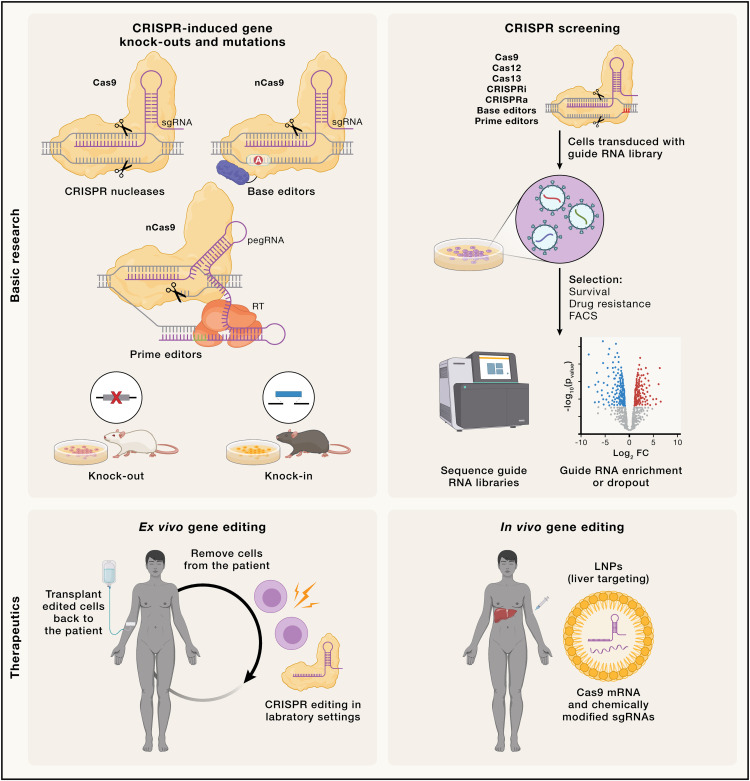
过去:CRISPR基因组编辑的发展与局限性
历史背景
基因组编辑的起源可以追溯到真核DNA修复领域的进展。20世纪90年代初的开创性研究表明,通过使用一种叫作I-SceI的同源内切酶,它可以识别18个碱基对的DNA序列,并在哺乳动物细胞中引入靶向的双链DNA断裂(DSB),从而促进目标位点的同源重组。这一发现奠定了利用DSB诱导来实现基因组编辑的基本概念。
为了开发可以识别长序列的核酸酶,研究人员设计了基于非特异性DNA内切酶与串联特异性DNA结合模块融合的工程化核酸酶。这些工具包括20世纪初推出的锌指核酸酶(ZFNs)和2010至2011年开发的转录激活因子样效应核酸酶(TALENs)。尽管这些方法为基因组编辑奠定了技术基础,但ZFNs和TALENs的设计与操作过于复杂,限制了它们的应用。
CRISPR关联(Cas)核酸酶的发现标志着基因组编辑技术的革命性转变。2007年,研究表明原核CRISPR-Cas系统是适应性基因组防御机制,能够识别并靶向病毒(如噬菌体)及其他移动遗传元件的外源核酸。这一系统通过捕获入侵DNA的片段并存储在重复阵列中生成CRISPR RNA(crRNA),这些crRNA作为分子向导,指导Cas蛋白识别并破坏外源核酸。
进一步的研究表明,II型CRISPR-Cas系统可以特异性地切割噬菌体DNA,其活性依赖于Cas9蛋白,并且需要一个额外的RNA成分,即转录激活crRNA(tracrRNA),来完成crRNA的成熟化。2012年发布的生化研究显示,Cas9是一种DNA切割的核酸内切酶,其特异性由由crRNA和tracrRNA组成的双RNA向导结构决定。为了简化操作,研究人员将tracrRNA和crRNA整合成单一向导RNA(sgRNA),从而提供了一种完全可编程的“一核酸酶-一向导RNA”设计。这一突破成为CRISPR基因组编辑技术的基础,通过设计与目标序列匹配的sgRNA,Cas9可以被精确靶向到特定的基因组位点。
RNA引导的Cas9切割DNA的发现引发了一场技术竞赛,研究人员争相将其改造成基因组编辑工具。最终,几项研究于2013年初发表,证明了在真核细胞中表达Cas9和特定的sgRNA能够在目标基因组位点引入遗传修改。
CRISPR核酸酶的基因组编辑功能
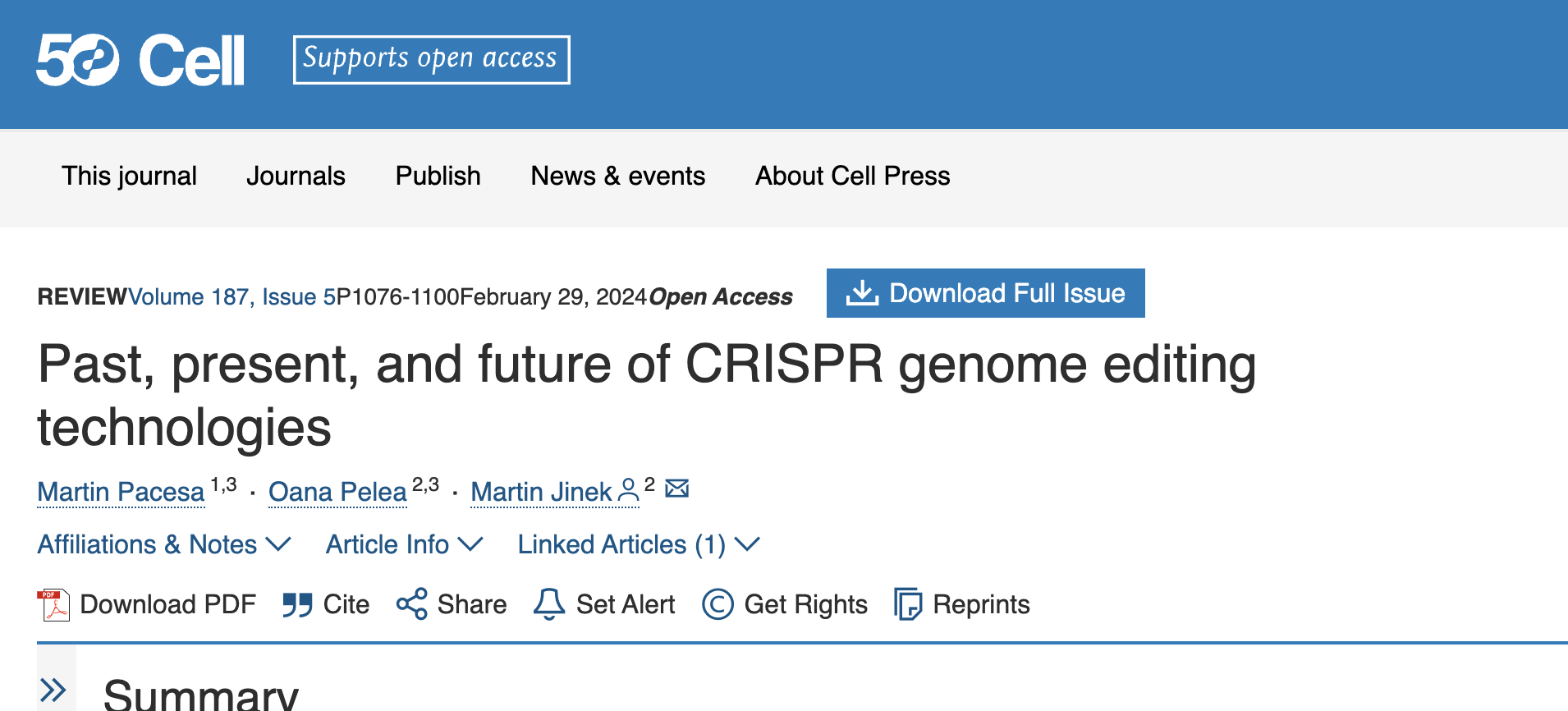
CRISPR-Cas核酸酶通过产生特异性的双链DNA断裂(DSB)迅速被改编为基因组编辑技术。第一个被改编用于基因编辑的Cas9蛋白来源于化脓性链球菌(SpCas9),由于其高活性和特异性,它仍然是应用最广泛的基因编辑工具。Cas9与crRNA-tracrRNA复合体或sgRNA结合后,形成具有活性的核酸内切酶。通过改变crRNA的5’端20个核苷酸的引导序列,Cas9可以被靶向到基因组的特定区域。
靶向结合还需要DNA非靶链(NTS)上紧邻目标位点下游的短原间隔区相邻基序(PAM)的存在。初步识别PAM后,Cas9会局部解链目标DNA,而向导RNA与目标链(TS)进行碱基配对,从PAM附近开始触发Cas9的构象变化并激活其核酸酶活性。
Cas9通过其HNH和RuvC结构域分别切割目标链(TS)和非目标链(NTS),在PAM上游3个核苷酸处产生DSB。通过选择性灭活其一个或两个核酸酶结构域,Cas9可以被改造成RNA引导的单链切割酶或纯粹的DNA结合蛋白,用于特定基因组位点的蛋白递送。
Cas12a的发现与功能
几年前,另一种核酸酶Cas12a(源自V型CRISPR-Cas系统)被发现并改编用于基因编辑。与Cas9不同,Cas12a不需要tracrRNA进行激活,而是通过识别crRNA的重复序列中的保守伪结结构进行自身引导RNA的核切加工。这一特点已被用于体内的多重编辑。
Cas12a靶向含有5’端TTTV PAM的DNA,并通过其单一RuvC结构域催化位于目标位点PAM远端的双链切割,生成5个核苷酸的5’突出端。切割后的DSB产物从蛋白中释放,而Cas12a仍保持催化活性,能够进一步切割单链DNA(ssDNA)。Cas12a被证明是一种高效的核酸酶,其精准的基因编辑能力可与Cas9互补。此外,Cas12a的转核酸酶活性还被用于序列特异性核酸检测。
传统基因组编辑方法与DSB修复机制
传统的基因组编辑方法依赖于在基因组中引入特异性的双链DNA断裂(DSB),随后通过细胞内的DNA修复机制进行修复。Cas9或Cas12a酶产生的DSB通常通过以下两种主要途径修复:
1.
末端连接修复路径(NHEJ或MMEJ)
:这是哺乳动物细胞中占主导地位的修复方式。该路径通过直接连接破损的DNA末端,通常导致小的插入或缺失(统称为indel),从而引起目标位点的蛋白编码序列破坏或基因敲除。此外,通过同时引入两个相邻的DSB,也可以删除特定的DNA片段。
2.
同源定向修复路径(HDR)
:HDR是一种精确的DSB修复途径,依赖于存在的同源DNA分子来指导修复结果。通过外源提供人工同源修复模板(如双链DNA或单链寡核苷酸ssODNs),HDR可以精确地在目标基因组位点引入期望的突变、插入或缺失。然而,HDR的效率通常受到多种因素的影响,例如修复模板的类型、递送方法、细胞类型和染色质环境。
尽管HDR在原则上能以核苷酸精度实现编辑,但其主要在细胞分裂活跃的S和G2期起作用,因此在非分裂细胞中的效率显著降低。
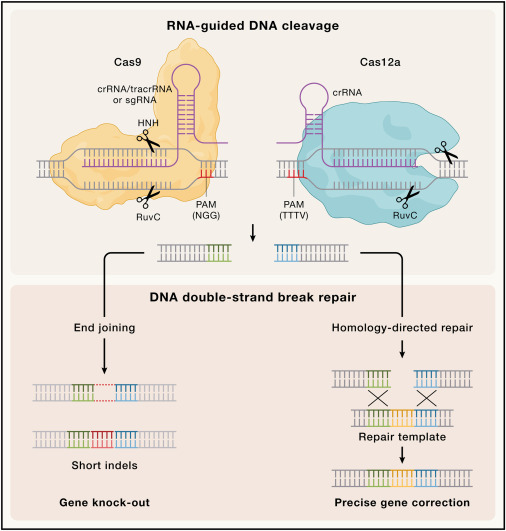
CRISPR基因组编辑的局限性
尽管CRISPR-Cas系统作为一种简单且有效的可编程基因编辑工具已极大地推动了基础研究和应用研究的发展,但其功能特点也带来了一些局限性。主要问题包括:
1.
脱靶效应
:自然的CRISPR-Cas系统在识别RNA向导与DNA目标之间的错配时具有一定的容忍性,这可能导致在基因组中未预期的部位发生编辑。
2.
靶向范围限制
:CRISPR核酸酶需要PAM序列的存在,这限制了其靶点选择范围。例如,SpCas9需要NGG的PAM序列,而Cas12a需要TTTV的PAM序列。
3.
依赖内源DSB修复机制
:由于NHEJ和HDR修复路径的竞争以及特定细胞类型中修复机制的差异,编辑结果可能不可控。
4.
递送挑战
:将CRISPR元件(例如Cas蛋白和导向RNA)高效递送到靶细胞或组织是基因编辑的一大障碍。递送载体的选择与目标细胞的特性密切相关。
脱靶效应的详细讨论
Cas9蛋白在一定程度上允许RNA向导与目标DNA之间存在错配,这一特性可能是其对抗高突变率噬菌体的一种进化适应。然而,在基因组编辑中,这种特性可能导致对部分互补的非目标位点的编辑。多项研究表明,Cas9能够容忍单个核苷酸错配甚至多个连续的错配,从而引发脱靶效应。
尽管如此,Cas9的脱靶效应在体外实验中更为常见,而在细胞中的发生率较低,这可能与基因组结构等因素有关。然而,在多个位点同时发生的脱靶编辑可能导致基因组重排,如缺失、倒位或染色体易位,并激活DNA损伤应激反应通路。为此,近年来针对脱靶效应的预测、检测和改进方法得到了大力开发,包括通过分子工程优化CRISPR基因编辑工具以提高其特异性。
靶向范围:PAM序列的限制
CRISPR核酸酶的DNA结合机制要求目标位点附近必须有PAM序列,这限制了其靶向范围。例如,最常用的SpCas9核酸酶需要NGG的PAM序列,理论上每8个核苷酸就可以找到一个目标位点,但在高A/T含量的基因组区域中,靶点选择会受到限制。
为了克服这一问题,自然界中具有不同PAM特异性的Cas9同源物被发现并应用于基因编辑。然而,这些Cas9变体往往对PAM的要求更为严格,从而限制了其应用范围。Cas12a核酸酶识别富含T的PAM序列(通常为TTTV),但同样面临靶向范围的限制。
近年来,研究人员开发了多种人工改造的Cas9变体,通过结构工程或定向进化改变其PAM特异性。这些变体极大地扩展了可靶向位点的范围,例如能够识别NGAN、NGCG等非G PAM序列,甚至开发了“几乎无PAM限制”的Cas9变体。然而,PAM靶向的放宽通常会降低编辑的特异性,并增加脱靶效应的风险。
控制编辑结果
通过靶向核酸酶在基因组中引入DSB显著提高了HDR修复率。然而,HDR的使用受到限制,仅在分裂细胞中有效,并且往往会因为末端连接修复路径的竞争而导致杂合性编辑结果。此外,编辑目标位点的精确性可能会受到以下不良结果的影响:
•
大规模的缺失和染色体重排
:DSB可能导致非目标DNA序列的大量缺失。
•
染色体丢失
:在某些情况下,DSB甚至可能导致整个染色体的丢失。
为提高HDR修复率,研究人员开发了多种方法,例如:
1. 使用非对称单链DNA寡核苷酸模板(ssODNs)来提高修复效率。
2. 引入沉默突变以阻止目标位点的反复切割。
3. 通过将修复供体模板与断裂位点结合来促进HDR。
此外,调控细胞周期(如延缓S期或增加G2/M期的比例)也能提高HDR的效率。近年来,一些研究还表明,将HDR相关修复因子(如Rad51、Rad52或Mre11)直接与Cas9结合,能够显著提高插入突变的效率。
尽管这些方法取得了一定的进展,但通过HDR引入长片段插入仍然充满挑战,尤其是在终末分化的细胞(如神经元)中,这类细胞HDR几乎不起作用。因此,为减少对DSB和HDR的依赖,研究人员开发了碱基编辑和引导编辑等新技术。
递送的挑战
基因编辑工具的递送仍然是体内和体外基因编辑应用的主要限制因素。尤其是在体内治疗中,将CRISPR元件安全、特异且高效地递送到目标细胞是成功基因编辑的先决条件。此外,CRISPR组件和递送载体的免疫原性也是体内应用的一个主要问题。研究表明,人类体内存在抗Cas9抗体和反应性T细胞,这可能会削弱治疗效果。
目前CRISPR元件的递送方法主要包括以下几种形式:
1.
体外递送
:在培养的细胞中,电穿孔(核转染)或脂质体介导的转染是最常用的递送方式,通常将Cas9和向导RNA以RNA、质粒DNA或预组装的核糖核蛋白(RNP)复合物的形式递送。
2.
体内递送
:常用病毒载体(如腺病毒、慢病毒和腺相关病毒AAV)将基因编辑模块递送至目标组织。其中,AAV因其低免疫原性、高转染效率和多样的细胞亲和性而受到青睐。然而,AAV的货物包装容量有限,无法一次性容纳较大的Cas9及其向导RNA。
除了病毒载体,脂质纳米颗粒(LNP)等非病毒递送方式也逐渐受到关注。这些方法相对更安全且免疫原性较低,但其效率和组织特异性仍需优化。
现在:当前技术及其应用
自CRISPR基因编辑技术首次问世以来,该领域以空前的速度发展。第一代依赖双链断裂(DSB)的基因组编辑工具(如基于Cas9和Cas12a核酸酶的编辑器)通过不断的技术创新,不仅增强了工具的多功能性,还提高了其精确性并最大限度地减少了非预期编辑的后果。然而,由于脱靶编辑活性和目标DSB可能引发的基因毒性(如p53激活)的担忧,该技术的安全性仍是主要问题。为此,研究人员开发了多种策略以实现CRISPR基因编辑工具的精确时空控制,例如利用天然存在的抗CRISPR蛋白抑制剂来实现组织限制性编辑。
此外,由于DSB的潜在基因毒性及HDR效率较低的问题,推动了“第二代”CRISPR技术的发展,这些技术无需依赖DSB和HDR即可实现基因组编辑,其中包括碱基编辑(BE)和引导编辑(PE)。这些新工具显著扩展了当前可用技术的范围,为不同类型的编辑任务和递送模式提供了量身定制的选择。尽管这些技术在克服传统CRISPR基因编辑限制方面取得了进展,但它们仍然面临活性、特异性和递送效率方面的限制。
Cas9之外的核酸酶
Cas9作为基因组编辑工具的开发激励了后续研究,科学家们努力寻找具有潜在基因组工程应用的新型天然Cas酶。在过去几年中,通过这些研究发现了十多种具有不同进化特性的RNA引导核酸酶,并被应用于基因编辑。
2015年Cas12a的发现标志着CRISPR领域的重要扩展。与Cas9相比,Cas12a核酸酶具有不同的PAM需求、向导RNA结构和DNA切割模式。研究表明,Cas12a在体内的特异性更高,脱靶活性更低,这部分归因于其较慢的DNA切割速率。此外,生物信息学分析发现了多种类型V Cas效应器,这些效应器在PAM和向导RNA需求方面具有独特特性,其中一些“最小化”的紧凑型核酸酶非常适合需要包装到小型病毒载体(如AAV)中的应用。
除了DNA靶向的Cas酶外,RNA靶向的核酸酶(如Cas13和Cas12g)也为分子编辑工具箱带来了新模式,允许对mRNA进行靶向降解或编辑,以及对核酸的高灵敏检测。
高保真Cas9变体
针对脱靶活性的问题,研究人员通过两种互补的方法开发了高保真SpCas9变体:
1.
结构设计
:通过结构工程消除Cas9蛋白与目标DNA之间的特定接触,增强向导RNA与目标DNA错配时的敏感性,从而减少脱靶结合和切割的可能性。
2.
定向进化
:利用进化方法筛选出能够减少脱靶编辑的Cas9突变体。
尽管这些变体在特异性方面显著优于野生型酶,但它们的编辑效率因目标DNA序列和应用场景而异。此外,由于每个目标位点的脱靶谱和编辑频率不同,目前尚无一种高保真酶可以普遍适用。
向导RNA的优化
通过优化向导RNA以提高特异性是一种具有吸引力的替代策略。例如:
• 缩短向导RNA的长度(例如从20个核苷酸缩短到17-18个核苷酸)可以显著降低脱靶活性,但同时可能降低效率或引入新的脱靶位点。
• 在向导RNA的5′端引入次级结构或未配对核苷酸,可以减弱脱靶效应。
• 使用化学修饰(如2′-O-甲基化或磷硫化修饰)可以增加向导RNA的稳定性和特异性,已在体内基因编辑中展现出良好效果。
扩展PAM特异性的编辑器
为克服PAM序列限制,研究人员通过结构工程和定向进化开发了识别非G PAM(如NGAN、NGCG)甚至几乎无PAM限制的Cas9变体。这些变体显著扩展了可靶向的基因组位点范围,同时简化了目标位点的选择过程。然而,这种扩展可能导致靶向特异性降低,并增加脱靶风险。
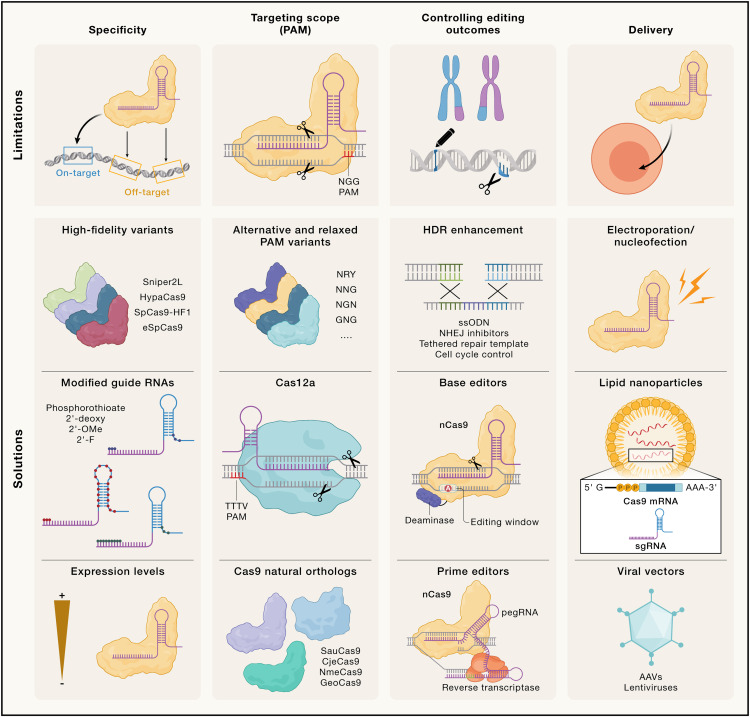
碱基编辑(Base Editing)
CRISPR衍生的碱基编辑技术是一种无需双链断裂(DSB)和同源修复模板的基因编辑工具,它可以高效地产生靶向点突变,因此在HDR缺乏的细胞中也能发挥作用。碱基编辑器(BE)是由一种失活的Cas9单链切割酶与核苷脱氨酶融合而成。
最初开发了两种主要的碱基编辑器:
1.
胞嘧啶碱基编辑器(CBE)
:由脱氨酶(如APOBEC1)和尿嘧啶糖苷酶抑制剂(UGI)结合,介导C-to-T(胞嘧啶转为胸腺嘧啶)的转换。
2.
腺嘌呤碱基编辑器(ABE)
:使用通过定向进化改造的腺苷脱氨酶(TadA),能够介导A-to-G(腺嘌呤转为鸟嘌呤)的转换。
Cas9引导碱基编辑器与目标DNA结合后,在非目标链上的编辑窗口内将胞嘧啶或腺嘌呤脱氨,分别产生尿嘧啶或次黄嘌呤。在复制或转录过程中,这些碱基被识别为胸腺嘧啶或鸟嘌呤,从而引入过渡性点突变。
近年来,CBE和ABE的多个改良版本显著提高了编辑活性,同时降低了脱氨酶诱导的脱靶编辑。这些编辑器还被扩展以覆盖A-to-C、A-to-Y和C-to-G等多种突变类型。由于碱基编辑结果可预测且精确,这项技术被广泛用于全基因组敲除筛选、突变研究,以及针对单点突变引发的疾病进行治疗性校正。然而,碱基编辑仍面临一些局限性,如低效率、旁侧编辑、不必要的编辑窗口过大以及潜在脱靶效应。
引导编辑(Prime Editing)
引导编辑是一种HDR独立的CRISPR编辑技术,能够产生靶向点突变、插入或删除。引导编辑器由以下两个主要部分组成:
1.
引导编辑RNA(pegRNA)
:包含与目标非靶链互补的3’端序列扩展,该序列带有预期的突变信息。
2.
融合蛋白
:由Cas9单链切割酶和逆转录酶(RT)结合而成。
引导编辑的过程如下:
1. Cas9切割非靶链,暴露的3’末端与pegRNA的模板序列配对。
2. 逆转录酶使用pegRNA作为模板,在目标位点引入突变。
3. 最终通过DNA链的重新退火和5’片段的修剪固定编辑。
该方法可在目标位点附近插入多达40个碱基或删除80个碱基,还可引入距离Cas9切割位点约30个碱基以内的点突变。改良后的引导编辑器进一步提高了编辑效率,包括增强pegRNA稳定性的结构改进、优化核定位信号,以及加入额外的向导RNA以促进编辑固定。然而,引导编辑的效率因目标序列和细胞类型而异,仍需进一步优化。此外,尽管引导编辑避免了常规DSB的产生,但可能仍会产生非预期的DSB和基因毒性。
转录调控:CRISPRi和CRISPRa
CRISPR技术不仅可以用于基因组编辑,还能够实现基因表达的瞬时调控:
1.
CRISPR干扰(CRISPRi)
:通过失活的Cas9(dCas9)结合KRAB转录抑制因子,靶向基因启动子区域,从而阻止RNA聚合酶转录,达到基因沉默的效果。
2.
CRISPR激活(CRISPRa)
:将dCas9与转录激活因子(如VP64)融合,用于激活特定基因的表达。通过组蛋白修饰酶(如去甲基化酶或乙酰转移酶)实现染色质状态的改变也可以达到激活基因的目的。
CRISPRi和CRISPRa技术已被广泛用于全基因组功能丧失和功能增益的筛选研究。然而,由于dCas9结合DNA的特异性低于其切割活性,因此这些技术的脱靶效应仍需改进。
RNA编辑和修饰
RNA编辑提供了瞬时且可逆的基因调控方法,特别适用于需要短期治疗效果的场景(如炎症或病毒感染)。目前基于Cas13的RNA编辑工具可以靶向降解RNA,或通过与RNA脱氨酶(如ADAR)融合来实现特定的碱基替换(如A-to-G)。此外,这些工具还被扩展为“表观转录组编辑器”,用于靶向修饰mRNA上的表观遗传标记(如N6-甲基腺苷)。
基础研究和人类医学中的应用
CRISPR基因编辑技术的进步带来了诸多应用,从基础研究到人类健康领域的治疗方法开发都有显著贡献。以下是一些重要应用实例:
1.
基础研究
:
•
疾病模型构建
:CRISPR技术使研究人员能够模拟疾病相关突变,从而在细胞、类器官以及动物模型中研究疾病的分子机制。例如,在患者来源细胞中引入特定突变或恢复突变至野生型,用以构建等位基因特异性疾病模型。
•
大规模基因筛选
:利用CRISPR进行全基因组敲除筛选,揭示特定基因在细胞生长、信号通路中的作用。
•
基因记录装置
:CRISPR系统被改造成分子记录工具,用于记录细胞分裂、转录活动或环境暴露的动态过程。
2.
临床应用
:
•
病毒基因组消除
:通过CRISPR技术靶向病毒基因组(如HIV或乙型肝炎病毒),抑制病毒复制。
•
CRISPR基因驱动
:在蚊子等媒介生物中引入不育性状,减少疾病传播(如疟疾)。
反向遗传学:疾病建模
通过CRISPR技术,研究人员可以创建等位基因特异性的疾病模型。例如:
•
细胞和类器官模型
:将人类诱导多能干细胞(iPSCs)通过CRISPR基因编辑转变为特定疾病相关的细胞类型(如神经元或心肌细胞),研究特定突变的功能。
•
动物模型
:CRISPR基因编辑技术被用于构建小鼠、斑马鱼、猪等动物模型,用以模拟人类疾病的病理过程。例如,CRISPR编辑的非人灵长类动物模型为发育过程和复杂疾病机制提供了新的见解。
CRISPR技术还能够实现多重基因编辑,例如通过多个向导RNA靶向不同基因位点,构建更复杂的疾病模型或实现大规模基因敲除。
正向遗传学:CRISPR筛选、谱系追踪与分子记录
CRISPR筛选
是研究健康与疾病中基因功能的强大工具。研究人员通过高通量筛选,可以系统性地扰动数千个基因,揭示与特定表型相关的基因。例如:
• 使用CRISPR核酸酶进行敲除筛选,识别参与癌症生长、药物耐受的基因。
• 利用CRISPRi和CRISPRa分别实现基因表达的抑制或激活,从而探索基因调控网络。
此外,结合单细胞组学技术(如单细胞RNA测序),CRISPR筛选使研究人员能够在单细胞水平上监测编辑引起的转录变化。研究还将这些方法应用于空间基因组学和肿瘤免疫微环境的研究。
谱系追踪
通过引入CRISPR编码的人工DNA条形码,可重建细胞分裂和发育的进程。例如,利用CRISPR在斑马鱼和小鼠胚胎中成功追踪了细胞的发育谱系,揭示了器官形成和癌细胞转移的动态过程。
体外治疗应用
CRISPR基因编辑的体外应用主要集中在细胞的基因修饰与回输治疗。以下是一些成功案例:
1.
血液疾病治疗
:通过在患者的造血干细胞中编辑BCL11A增强子,重新激活胎儿血红蛋白表达,治疗镰刀型细胞病(SCD)和β地中海贫血(TDT)。
2.
嵌合抗原受体(CAR)T细胞疗法
:通过CRISPR编辑T细胞的特定位点插入CAR基因,避免传统病毒载体引发的潜在安全性问题。此外,通过多重基因敲除提高CAR-T细胞的持久性和功能。
体外编辑的优势在于能够精确控制编辑效率并减少体内免疫反应。然而,如何提高编辑效率、降低脱靶效应,仍是体外治疗中的重要挑战。
体内治疗应用
体内基因编辑涉及直接将CRISPR元件递送到患者体内以修复受损基因或敲除致病基因。例如:
1.
肌肉疾病治疗
:通过AAV递送Cas9和向导RNA,恢复杜氏肌营养不良症(DMD)动物模型中的肌肉功能。
2.
视网膜疾病治疗
:通过亚视网膜注射,将CRISPR系统递送至眼部,修复CEP290基因突变以治疗Leber先天性黑蒙。
3.
肝脏疾病治疗
:利用脂质纳米颗粒(LNP)将CRISPR递送至肝细胞,用于敲除转甲状腺素基因(TTR),显著降低血清TTR水平。
这些体内治疗方法在临床试验中展现出良好的前景,尤其是在对肝脏和眼部等易于递送的组织中。然而,对于更难以到达的组织和器官,递送系统仍需进一步改进。
未来:新兴技术与展望
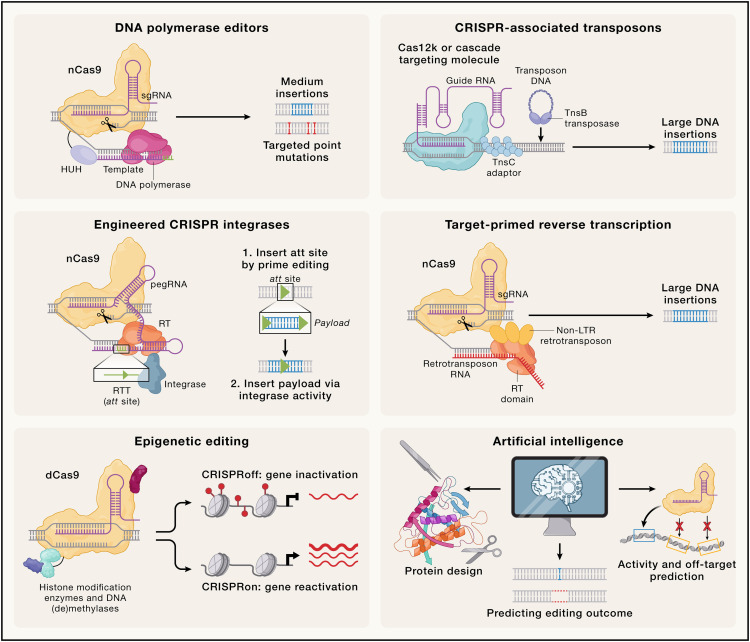
随着当前CRISPR技术局限性的逐步显现,研究人员正不断开发和完善新的方法,以提高CRISPR技术的效率和多样性。这些新兴技术(第三代CRISPR工具)包括以下几个重要方向:
RNA引导的紧凑型核酸酶
开发新型RNA引导核酸酶的主要目的是寻找具有更小分子量、更广靶点适应性和更高效率的编辑工具。这些核酸酶有助于解决递送难题,并可与其他功能模块(如碱基编辑器和引导编辑器)结合,用于更广泛的应用。
近期的研究发现了一些紧凑型核酸酶:
1.
Cas12f
:仅有422个氨基酸,通过深度突变扫描和结构导向设计增强了其体内编辑活性。
2.
IscB/TnpB
:这些属于CRISPR早期进化的前体蛋白,天然存在于原核转座元件中,能够在真核细胞中实现可编程的基因组编辑。
3.
Fanzor
:一种真核TnpB类似蛋白,具有RNA引导的DNA切割活性,其紧凑性使其在病毒载体递送中具备优势。
尽管这些核酸酶提供了更高的递送灵活性,但其编辑效率和靶点范围仍需进一步优化。
DNA聚合酶编辑器
DNA聚合酶编辑器是另一种无需依赖HDR的基因编辑工具,它通过在目标DNA位置引入多种突变或较长插入来实现编辑。例如:
•
引入随机突变
:通过将一种误差倾向型DNA聚合酶与Cas9融合,可以在目标位置附近实现350个核苷酸范围内的多样化编辑。
•
大规模插入
:使用线性DNA模板和逆转录酶结合Cas9切口编辑,能够在目标位点插入超过100个核苷酸的序列。
这些技术的优点在于其编辑结果的多样性和精确性,同时避免了DSB可能带来的基因毒性。
CRISPR引导的重组酶与转座酶
基于DSB的编辑在非分裂细胞中效率较低,而重组酶和转座酶技术提供了一种无需依赖DSB即可插入长DNA片段的潜力。相关研究包括:
1.
CRISPR-重组酶系统
:通过在目标位点引入特定识别序列,结合重组酶实现长片段插入。
2.
CRISPR转座酶系统(CAST)
:天然存在的I型或V型CRISPR-Cas系统结合转座元件,可用于细菌中的高效DNA插入。
尽管这类技术在细菌中表现出良好效果,但在哺乳动物细胞中的效率仍需优化。
反转录转座子(Retrotransposons)
基于反转录的转座子技术可以实现长DNA片段的高效插入。这些转座子通过靶向引导的逆转录作用,将RNA转录的信息整合到目标DNA中。最新研究表明,通过与Cas9融合,转座子可以被重新靶向至非原生DNA位点。这种方法可能比其他技术更适合大基因片段的插入。
表观基因组编辑
CRISPR技术不仅可以进行基因组修改,还可以用于改变基因的表观遗传状态。表观基因组编辑通过引入可逆的表观遗传修饰(如DNA甲基化、组蛋白乙酰化)来调控基因表达:
1.
CRISPRoff/CRISPRon
:通过DNA甲基化酶和去甲基化酶组合,实现基因表达的持久抑制或激活。
2.
组蛋白修饰
:利用乙酰化转移酶或去乙酰化酶调整染色质状态,诱导或抑制目标基因的表达。
这些方法可以实现非永久性的基因表达调控,避免引入不可逆的基因组改变。
RNA编辑
相较于DNA编辑,RNA编辑提供了一种更安全、瞬时的基因调控方式。相关技术包括:
1.
Cas13 RNA编辑器
:通过与脱氨酶结合,可实现A-to-G或C-to-U的RNA编辑。
2.
长RNA剪接与替换
:通过RNA靶向的Cas13与剪接RNA的结合,可插入或替换mRNA的大段序列。
RNA编辑的瞬时性和可逆性使其在治疗急性疾病(如炎症、病毒感染)中具有广阔前景。
新型递送方法
递送效率和特异性仍是CRISPR基因编辑的主要瓶颈。为解决这一问题,研究人员开发了多种新递送方法:
1.
脂质纳米颗粒(LNP)
:优化配方以实现组织特异性递送。
2.
细胞穿透肽
:用于编辑神经元和免疫细胞,具有低免疫原性。
3.
病毒样颗粒(VLPs)
:利用天然病毒样结构,递送CRISPR组件而不引入病毒遗传物质。
尽管这些方法展现出良好前景,但其在体内的长期安全性和扩展能力仍需进一步验证。
人工智能的应用
人工智能(AI)正迅速改变基因编辑领域,通过预测编辑效率与脱靶风险,显著提高了编辑工具的精确性。AI还被用于设计新型高效核酸酶以及优化递送系统。然而,AI模型的训练依赖于高质量的数据,并且其“黑箱”特性限制了模型决策的可解释性。
展望
CRISPR技术正从第一代DSB依赖的工具逐步扩展到第二代(碱基编辑、引导编辑)及第三代(重组酶、转座酶)的技术。这些工具不仅提高了基因编辑的精确性和安全性,还扩大了其应用范围。未来,基因组编辑的核心挑战将集中在递送系统的改进以及编辑效率和脱靶效应的进一步优化。
尽管存在伦理与社会争议,特别是关于人类生殖系基因编辑的讨论,CRISPR技术仍有望在医学、农业和生态学领域产生深远影响,推动人类社会的可持续发展。
回复
支持
反对
举报
返回列表
发表回复
高级模式
B
Color
Image
Link
Quote
Code
Smilies
您需要登录后才可以回帖
登录
|
立即注册
本版积分规则
发表回复
回帖后跳转到最后一页
关闭
官方推荐
/3
AI助手<小桔子>来了!
欢迎来交流,可以回答IVD行业各类问题!
查看 »
IVD业界薪资调查(月薪/税前)
长期活动,投票后可见结果!看看咱们这个行业个人的前景如何。请热爱行业的桔友们积极参与!
查看 »
小桔灯网视频号开通了!
扫描二维码,关注视频号!
查看 »
返回顶部
快速回复
返回列表
客服中心
搜索
洽谈合作
关注微信
微信扫一扫关注本站公众号
个人中心
个人中心
登录或注册
业务合作
-
投稿通道
-
友链申请
-
手机版
-
联系我们
-
免责声明
-
返回首页
Copyright © 2008-2024
小桔灯网
(https://www.iivd.net) 版权所有 All Rights Reserved.
免责声明: 本网不承担任何由内容提供商提供的信息所引起的争议和法律责任。
Powered by
Discuz!
X5.0 技术支持:
宇翼科技
浙ICP备18026348号-2
浙公网安备33010802005999号
快速回复
返回顶部
返回列表

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 






 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 发表于 2024-12-25 14:34
发表于 2024-12-25 14:34