金桔
金币
威望
贡献
回帖0
精华
在线时间 小时
|
本文内容摘自美国FDA对于FDA检查的官方网站,数据内容来自美国FDA对于检查结果的数据库
FDA检查简介
FDA非常认真地履行其责任,确保我们所食用的食品安全,我们使用和依赖的医疗产品符合我们严格的质量、安全和有效性标准。该机构使用基于风险的方法来确定需要检查的国内外设施。
检查是一种仔细、批判性的官方现场检查,旨在确定设施是否符合联邦法律规定。尽管检查对于FDA的监督至关重要,但它们只是时间的一瞬间,这就是为什么检查只是监督FDA监管产品安全和质量的全面方法的一部分。
在美国以外生产的产品必须符合与在美国制造的产品相同的标准。该机构对全球供应链中的潜在问题保持警惕,以确保美国人无论产品在哪里制造,都对其产品的安全和质量充满信心。
FDA检查的内容包括:
- 疫苗和药品制造商
- 血库
- 食品加工设施
- 奶制品和农产品种植场
- 动物饲料加工厂
- 外包设施和复方药房
- 烟草制造商
FDA还会检查:
- 进行人体研究(临床试验)的设施
- 进行动物或微生物研究的实验室,当这些研究用于申请FDA批准医疗产品时
- 销售在美国的FDA监管产品的国外制造和加工场所
- 在边境检查进口的受管制产品
FDA 检查的类别
FDA进行多种类型的检查,以帮助提供所需的医疗产品,并保护消费者和患者免受不安全产品的伤害:
监测检查
旨在监控市场上的制造过程和FDA监管产品的质量。机构利用检查评估制造商是否符合质量制造规范。
因果检查
是在机构有理由相信设施存在质量问题时触发的,以跟进投诉或评估纠正措施以解决先前的违规行为。
基于申请的检查
占机构进行的申请审查的约20%。这些检查是市场推广新药物、设备或生物制品的申请审查过程的一部分,以确定新产品是否符合FDA法规的制造,并确保设施能够持续生产该产品,并且提交的数据准确完整。此外,机构还对烟草设施进行检查,作为烟草申请的市场前评估的一部分。
机构还进行检查,以验证待
处理申请所支持的临床和非临床研究的可靠性、完整性和合规性。
检查可以是对设施运营的全面审查,也可以是针对特定问题或问题的定向检查,有时称为有限检查,例如确保符合召回行动或跟进设施的纠正措施。
风险导向的检查方法
FDA使用基于风险的评估来选择外国和国内的医疗产品制造设施进行检查。机构根据一系列具体的标准,如设施类型(制造商、控制实验室等)、设施的合规历史(包括是否在过去四年内进行过检查)、危险信号(包括与设施相关的召回记录、历史和性质等)、设施生产的产品的固有风险(如剂型、给药途径、用于灭菌的产品、剂型中活性药物成分的浓度、紧急使用等)以及设施是否接受过外国监管合作伙伴的检查等,对被视为高风险的医疗产品监测检查进行优先考虑。例如,一个以前未经过检查并生产狭窄治疗指数药物的无菌药物制造场地,可能被认为比一个具有良好的检查和合规历史,生产非处方固体口服剂型药物的场地风险更高。
对于生物研究监测(临床和非临床研究)的检查,机构考虑了几个基于风险的因素,包括:
合规历史
与该场地相关的投诉
场地关联的申请数量
招募人数
协议异常/偏差数量
数据异常
人类和动物食品的检查受到《食品安全现代化法案》检查频率要求的驱动,要求对国内高风险设施进行每三年一次的检查,对非高风险设施进行每五年一次的检查。
FDA 检查分类
我们如何对检查进行分类
在进行检查后,机构评估检查结果,确定设施是否符合适用的法律和法规,并对检查进行分类。
有三种分类:
- 无需采取行动(NAI),意味着在检查过程中没有发现任何不良条件或做法,
- 建议采取自愿行动(VAI),意味着发现了不良条件或做法,但机构未准备采取或建议任何行政或监管行动,
- 建议采取正式行动(OAI),意味着建议采取监管和/或行政行动。
该机构维护一个检查数据仪表板,其中包括http://FDA.gov上每周更新的最终检查分类。一旦对检查进行了分类,并且被认为已关闭,它将被添加到此数据库中。
然而,并非所有检查都包含在数据库中。不包括在内的有:
由各州进行的检查,批准前检查,乳腺X射线设施检查,等待最终执法行动的检查以及非临床实验室的检查。
非临床实验室的检查可在符合良好实验室规范要求的非临床实验室中进行。
机构通过评估检查期间收集的信息以及检查后设施提供的信息来确定最终的检查分类。调查员对检查分类的建议是评估和分类过程中的重要因素。还会考虑其他信息来确定最终的检查分类,包括设施对在FDA 483表格中突出显示的检查观察结果的回应以及公司提出的或已完成的纠正措施。
FDA 对中国公司的检查
下面的数据结果是我从美国FDA检查结果数据库重获取的信息,这里给出了自2009年至2023年11月底,美国FDA对于中国地区出口到美国需要经过FDA 审查的公司的检查汇总。
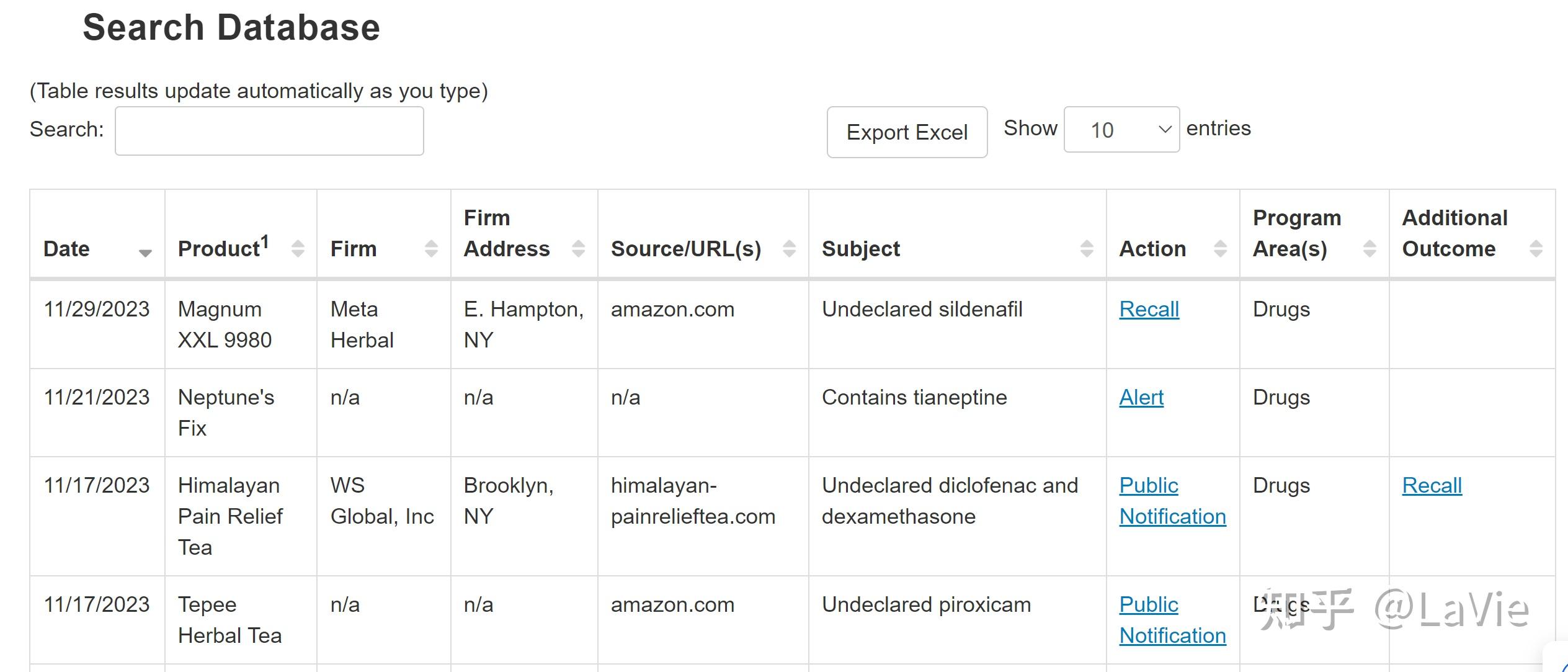
第一幅图可以看出自疫情开始,FDA对中国地区的检查已经大幅降低,从2019年之前的300-400,减少到了100以下。疫情之后,FDA对中国地区的审查也可能会增加。
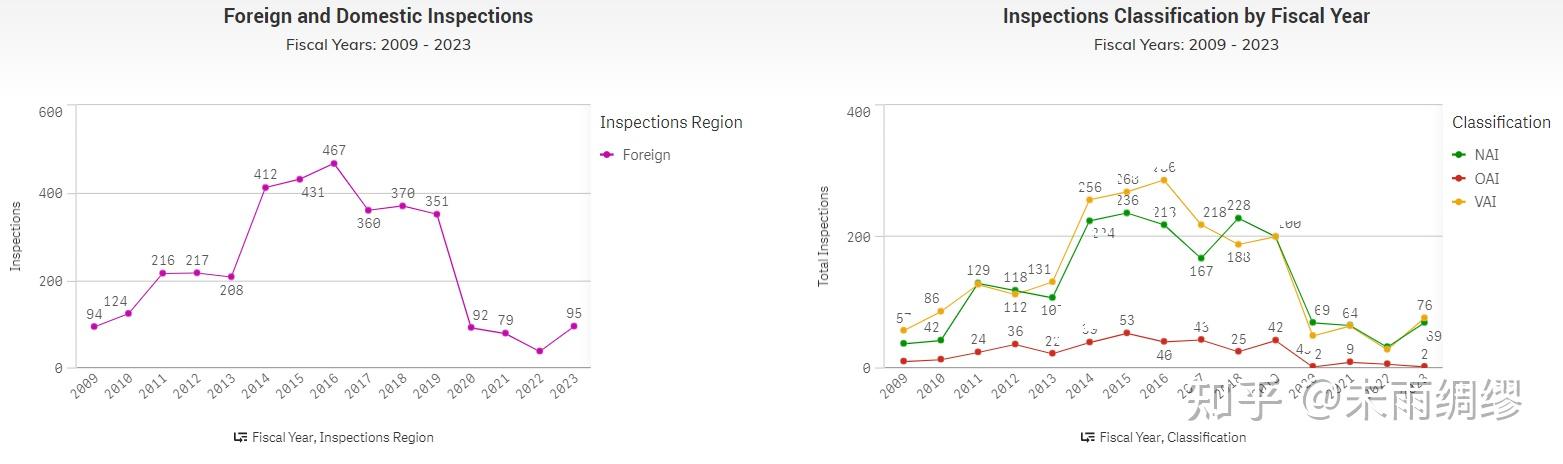
结果来自于美国FDA检查结果数据库
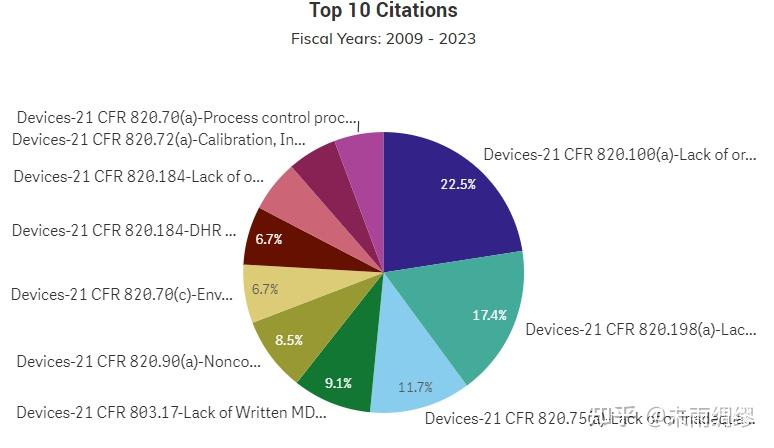
结果来自于美国FDA检查结果数据库
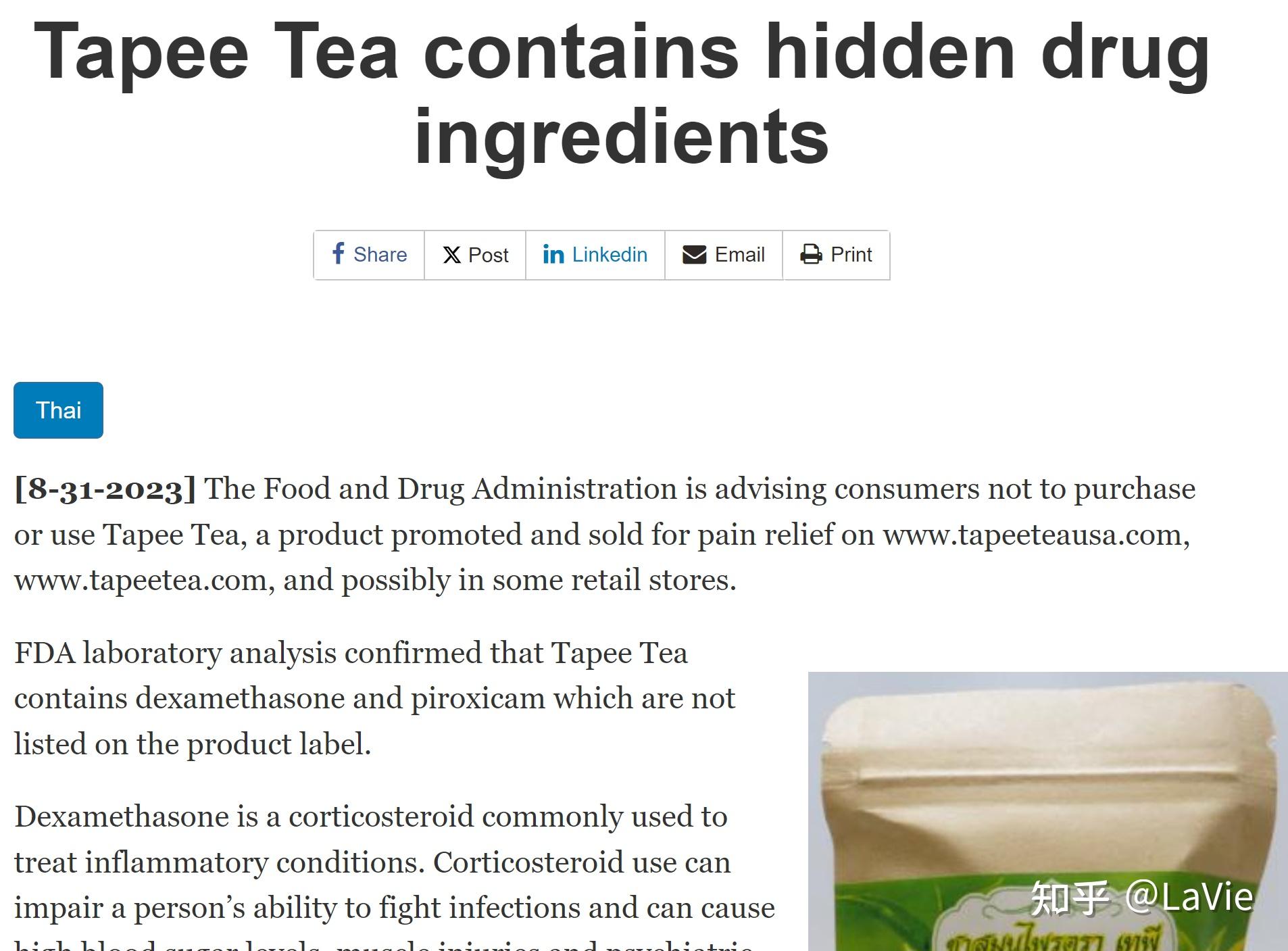
第二幅图的结果是,在以上筛选的的审查结果中,FDA对于不合规或者建议整改的措施中,都引用了FDA法规中的哪些条例,可以看出前三名的条例分别是:
- 21 CFR 820.100 Corrective and Preventive Action (a) Each manufacturer shall establish and maintain procedures for implementing corrective and preventive action. The procedures shall include requirements, (21 CFR 820.100 纠正和预防措施 (a) 每个制造商都应建立和维护纠正和预防措施的实施程序。该程序应包括要求,这条占22.5%)
- 21 CFR 820.198 Complaint files (a) :Each manufacturer shall maintain complaint files. Each manufacturer shall establish and maintain procedures for receiving, reviewing, and evaluating complaints by a formally designated unit. Such procedures shall ensure that: (1) All complaints are processed in a uniform and timely manner; (2) Oral complaints are documented upon receipt; and (3) Complaints are evaluated to determine whether the complaint represents an event which is required to be reported to FDA under part 803 of this chapter, Medical Device Reporting. (21 CFR 820.198 投诉文件 (a):每个制造商都应保留投诉文件。每个制造商都应建立和维护接收、审核和评估投诉的程序,并指定一个正式的单位负责。此类程序应确保:(1) 所有投诉以统一和及时的方式处理;(2) 口头投诉在接收时进行记录;以及(3) 评估投诉以确定投诉是否代表根据本章节第803部分《医疗器械报告》的要求需向FDA报告的事件。这条占17.4%)
- 21 CFR 820.75 Process validation (a) Where the results of a process cannot be fully verified by subsequent inspection and test, the process shall be validated with a high degree of assurance and approved according to established procedures. The validation activities and results, including the date and signature of the individual(s) approving the validation and where appropriate the major equipment validated, shall be documented. (21 CFR 820.75 过程验证 (a) 如果后续的检查和测试无法对过程的结果进行完全验证,该过程应经过高度保证的验证,并按照已建立的程序获得批准。验证活动和结果,包括批准验证的个人的日期和签名,以及适当的情况下经过验证的主要设备,应进行记录。这条占11.7%)
|
|

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 






 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 发表于 2024-12-11 13:12
发表于 2024-12-11 13:12


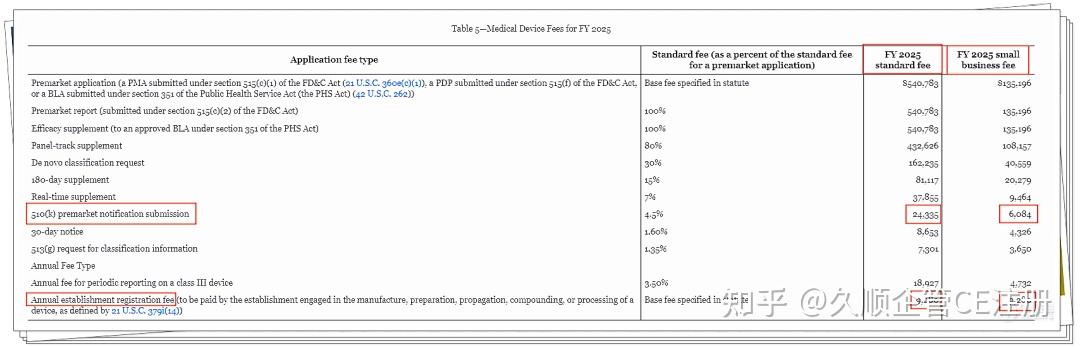
 发表于 2024-12-11 13:13
发表于 2024-12-11 13:13

 发表于 2024-12-11 13:14
发表于 2024-12-11 13:14



