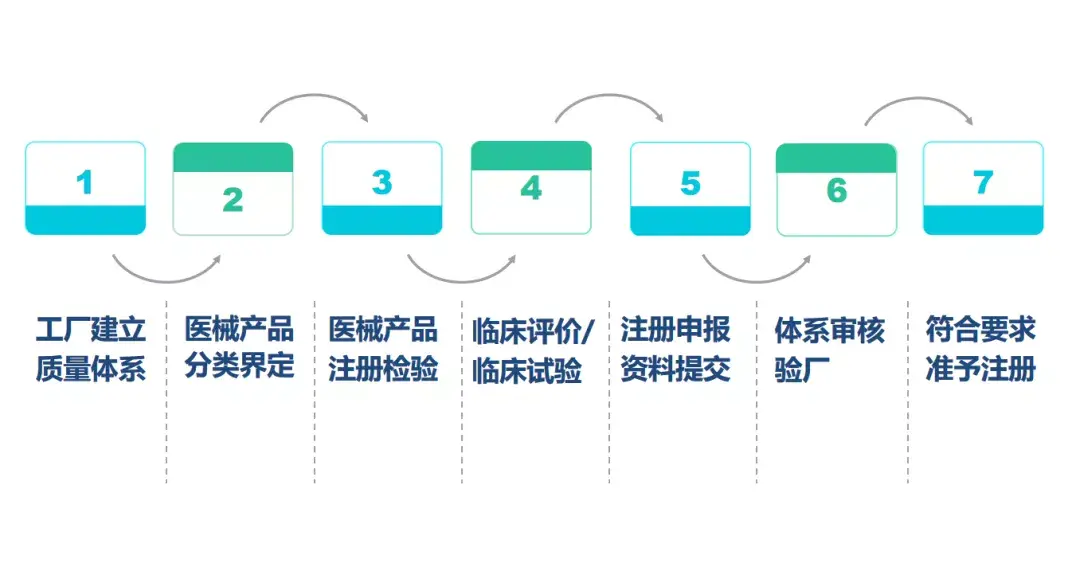
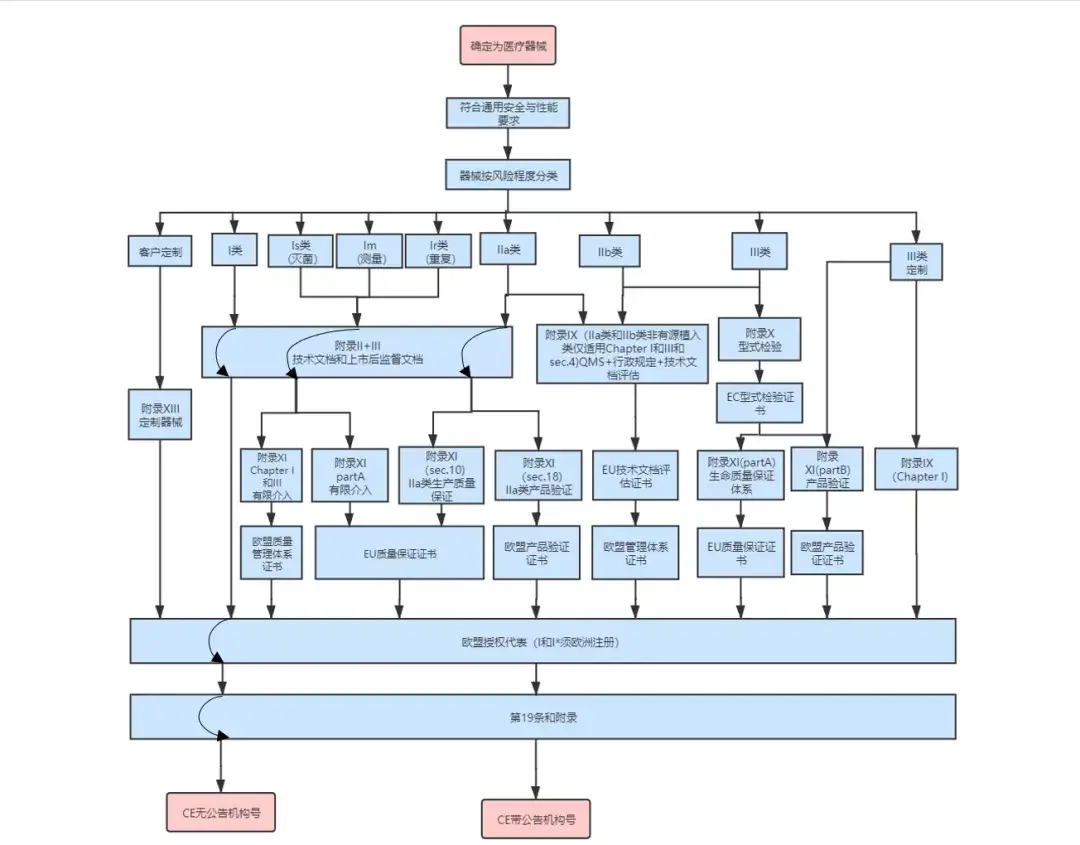
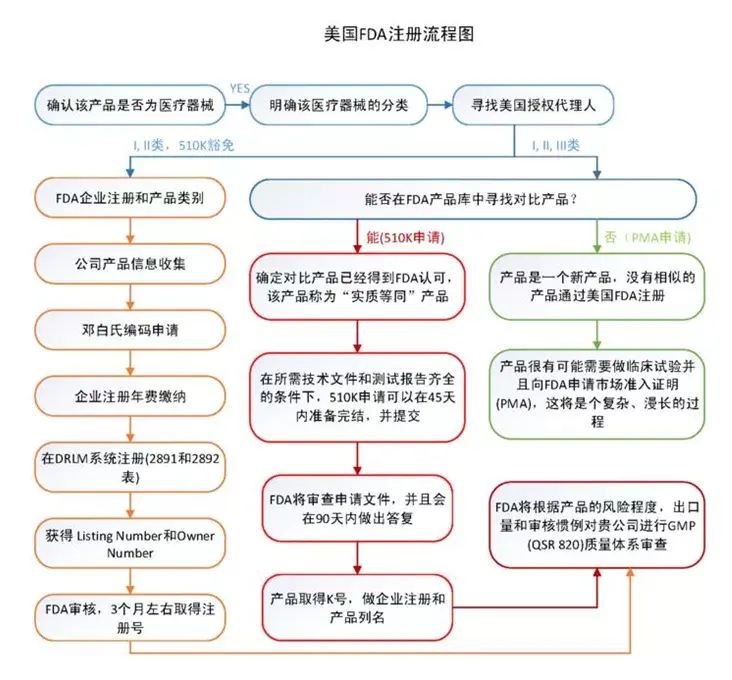
3、 美国FDA认证
FDA认证即美国食品药品监督管理局(Foodand Drug Administration,简称FDA)为确保美国本国生产或进口的食品、化妆品、药物、生物制剂、设备和放射产品的安全而设立的审查机制,在美国等近百个国家,只有通过了FDA认可的材料、器械和技术才能进行商业化临床应用。根据风险等级的不同,FDA将医疗器械分为三类(Ⅰ,Ⅱ,Ⅲ),其中Ⅲ类风险等级最高,风险等级越高则监督越多。FDA明确规定了每一种医疗器械的产品分类和管理要求,FDA医疗器械产品目录已收录超过1700多种产品。医疗器械FDA注册类型包括:

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 






 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 发表于 2024-12-10 22:57
发表于 2024-12-10 22:57