金桔
金币
威望
贡献
回帖0
精华
在线时间 小时
|
登陆有奖并可浏览互动!
您需要 登录 才可以下载或查看,没有账号?立即注册


×
今天为大家介绍的是来自Remo Rohs团队的一篇论文。预测蛋白质与DNA结合特异性是理解基因调控中一个既具挑战性又至关重要的任务。蛋白质-DNA复合物通常会与特定的DNA靶位点结合,而一个蛋白质可以以不同的结合特异性与多种DNA序列结合。这些信息无法通过单一结构直接获得。为了解决这一问题,作者提出了结合特异性深度预测模型(DeepPBS),这是一个几何深度学习模型,旨在从蛋白质-DNA结构中预测结合特异性。DeepPBS可以应用于实验获得的结构或预测的结构。可以提取出界面残基的可解释蛋白质重原子重要性评分。这些评分汇总到蛋白质残基层面,已通过了诱变实验的验证。在设计用于特定DNA序列的靶向蛋白时,DeepPBS被证明能够预测出实验测量的结合特异性(即跟实验测量一样准确)。DeepPBS为机器辅助研究提供了基础,这些研究可以推动我们对分子相互作用的理解,并指导实验设计和合成生物学的研究。

转录因子在各种调控功能中起着关键作用,这些功能对于生命的各个方面都至关重要。因此,理解蛋白质如何靶向特定DNA序列的机制非常重要。广泛的研究揭示了众多导致高特异性结合的机制,包括精氨酸残基在DNA小沟中的强静电相互作用、脱氧核糖与苯丙氨酸的堆叠、大沟中鸟嘌呤与精氨酸之间的双齿氢键以及其他相互作用。
通常,蛋白质-DNA结构通过X射线晶体学、核磁共振光谱或冷冻电子显微镜实验获得,并存储在蛋白质数据库(PDB)中。这些结构通常展示了一个结合的DNA序列及其相关的物理化学相互作用,但并未涵盖所有可能的结合DNA序列。相反,这些信息可以通过蛋白质结合微阵列、结合高通量测序的系统进化配体富集技术(SELEX-seq)、染色质免疫沉淀后测序、高通量SELEX或相关的高通量方法来实验获取。这些实验捕获了可能结合的DNA序列范围,但不一定提供结构信息。实际上,这些实验是互补的,通常需要人工检查以将结构数据中的分子相互作用细节与结合特异性数据进行关联。
尽管在某些特定蛋白质家族的研究中取得了一定进展,但在整个蛋白质家族中预测给定蛋白质序列的结合特异性仍然是一个具有挑战性且未解决的问题。结合过程中结构的变化,以及机制上的巨大多样性,使得这一问题更加复杂。蛋白质-DNA结构中包含了宝贵的信息,人工智能可以利用这些信息实现跨蛋白质家族的普适性。
模型架构
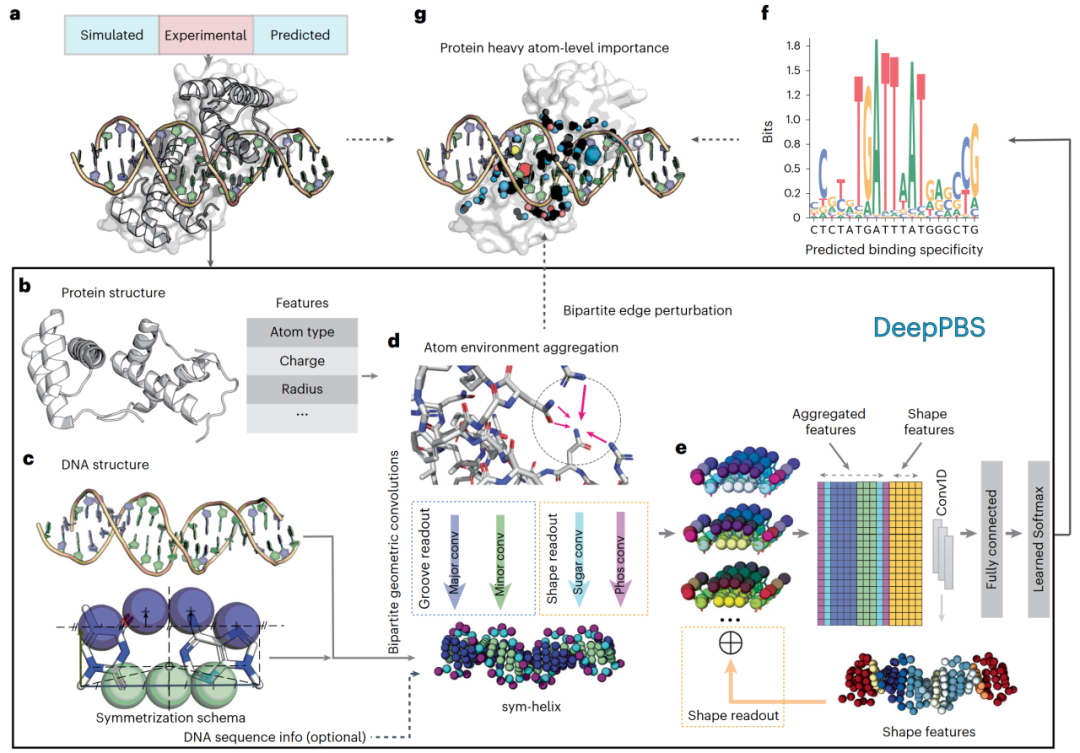
图 1
图1展示了DeepPBS框架的工作流程。DeepPBS的输入(图1a)包括一个蛋白质-DNA复合物结构,其中一条或多条蛋白质链与DNA双螺旋结合。这类结构的潜在来源包括实验数据(例如,PDB)、分子模拟快照或设计的复合物。DeepPBS将该结构处理为一个二分图,蛋白质和DNA部分分别采用不同的空间图表示。蛋白质图是一个基于原子的图,重原子作为顶点。在这些顶点上计算了多个特征(图1b)。DNA则被表示为对称螺旋(sym-helix),这一表示方式在保留双螺旋形状的同时去除了DNA的序列特征。如有需要,还可以选择将DNA序列信息重新引入为对称螺旋点的特征。
DeepPBS在蛋白质图上执行一系列空间图卷积,以聚合原子邻域信息(图1d)。DeepPBS的下一个关键组成部分是一组从蛋白质图到对称螺旋的二分几何卷积(图1d)。特定的化学相互作用(例如氢键)依赖于位置和方向。DeepPBS学习对称螺旋点的几何方向如何与邻近蛋白质残基的方向和化学性质相关联。针对对称螺旋点,采用了四种不同的二分卷积,分别对应大沟、小沟、磷酸基团和糖基团。大沟和小沟的卷积被称为“沟槽读出”(groove readout),这个术语取代了“碱基读出”,因为对称螺旋中去除了碱基的标识。磷酸基团和糖基团的卷积结合DNA形状信息,形成“形状读出”(图1e)。“沟槽读出”和“形状读出”因素共同决定了不同蛋白质家族在不同程度上的结合特异性。在此基础上,对称螺旋表示可以将聚合的三维对称螺旋特征简化为一维的碱基对级别特征。通过添加DNA形状信息并实施一维卷积神经网络和预测层(图1e),DeepPBS最终预测结合特异性(图1f)。
由于现有文献中没有针对从蛋白质-DNA复合物结构数据预测跨蛋白质家族结合特异性的标准数据集,作者需要建立一个数据集用于交叉验证和基准测试。
DeepPBS在经实验确定的结构上的表现
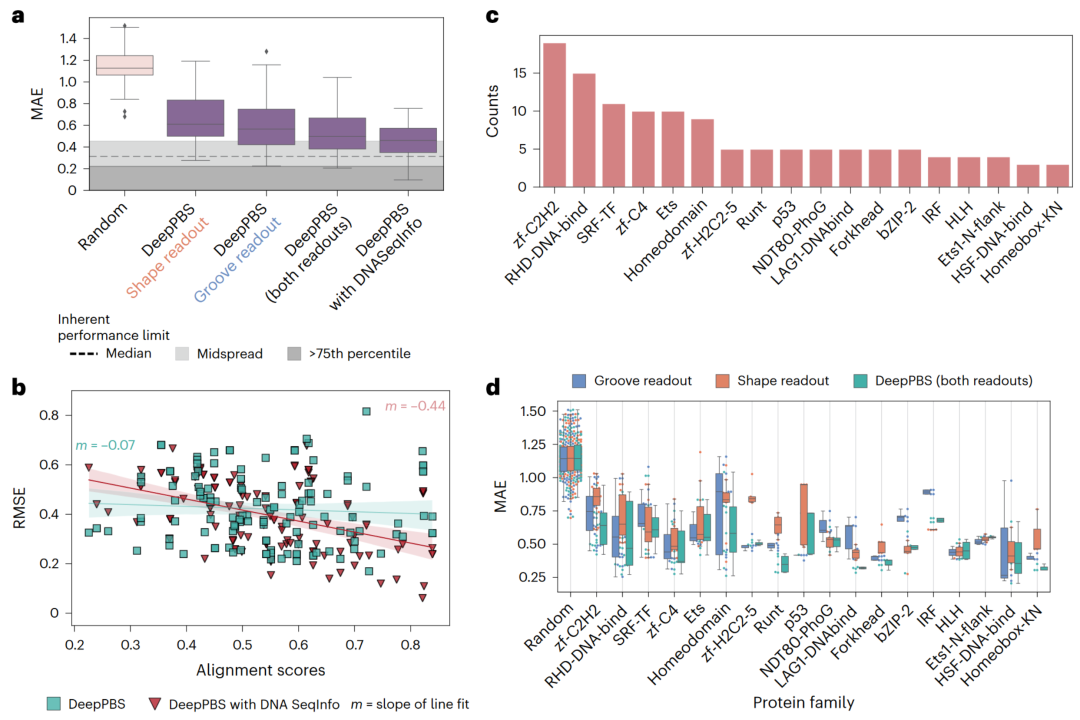
图 2
作者使用DeepPBS集成模型对基准数据集进行了评估。DeepPBS架构允许模型通过两种机制进行训练:“沟槽读出”不涉及主链卷积并排除形状信息,而“形状读出”则不涉及沟槽卷积(图1d, e)。DeepPBS(同时执行“沟槽读出”和“形状读出”模式)和这两种变体的基准性能如图2a所示。中位性能方面,“沟槽读出”版本优于“形状读出”版本,而DeepPBS模型在这两种模式各自独立时表现更好(双侧t检验P值<0.01;图2a)。
数据集由实验确定的结构构建,因此,共晶结构衍生的DNA序列通常是一个合理的结合序列示例。如预期的那样,将序列信息整合到对称螺旋点中(“DeepPBS with DNASeqInfo”)提高了性能(图2a),显著缩小了数据集中固有性能极限的差距。这一极限源于用于创建数据集的两个数据库在同一蛋白质的结合特异性数据上可能存在一定程度的不一致性。作者计算了在两个数据库中都出现的所有唯一PWM的不一致性分布。然而,从可解释性和设计的角度来看,特别是在结合的DNA序列可能不具代表性的情况下,“DeepPBS”模型由于对结构中的DNA序列敏感性较低,因此是最佳选择。这一事实通过比较“DeepPBS”和“带有DNA SeqInfo的DeepPBS”模型在PWM与共晶结构衍生的DNA比对得分中的表现得到验证。与包含DNA序列信息的变化拟合线相比(均方根误差(RMSE)的斜率为−0.44,平均绝对误差(MAE)的斜率为−0.62),DeepPBS预测的拟合线斜率更接近于零(图2b)。
DeepPBS捕捉到了家族特异性结合模式的规律
基准集中的不同蛋白质家族的丰度如图2c所示(交叉验证集的丰度见补充图5b)。家族注释来源于蛋白质家族数据库(PFAM)。该数据集涵盖了多种DNA结合蛋白家族。DeepPBS在各种蛋白质家族中的表现提供了几个关键见解。DeepPBS展示了在各个蛋白质家族中的良好普适性,即使在结构相对较少的家族中也表现出色(图2d),如热休克因子蛋白。这一观察结果表明,该模型学习的是蛋白质-DNA结合的基本机制,而不是在家族特异性模式上过拟合。
进一步的验证来自比较DeepPBS“沟槽读出”和“形状读出”模型的表现(图2d)。对于zf-C2H2和zf-C4等家族,“形状读出”模型的表现不如“沟槽读出”模型。这一结果与这些家族已知的结合机制一致。例如,zf-C2H2使用锌指结构域扫描DNA以寻找合适的碱基相互作用,过程中DNA的弯曲或构象变化较小。这种结合模式使得zf-C2H2家族成为基于蛋白质序列的结合特异性预测和设计的常见目标。相反,像干扰素调节因子(IRF)蛋白(图2d)和T-box蛋白等家族,“形状读出”模型表现更好,这与其已知的结合机制一致,这些机制涉及显著的构象变化。对于同源域(HD)和叉头蛋白等家族(图2d),DeepPBS模型的表现优于“沟槽读出”和“形状读出”两个组件。这一结果表明,网络捕捉到了这些组件的复杂高阶关系。
应用于计算预测的蛋白质-DNA复合物
DeepPBS框架不仅限于实验结构。随着人工智能推动的可扩展结构预测方法的最新进展,提供了前所未有的潜力。具体而言,像RFNA和MELD-DNA这样的模型可以用于从序列预测蛋白质-DNA复合物的结构。这类预测算法为DeepPBS应用于缺乏实验DNA结合结构数据的蛋白质铺平了道路。
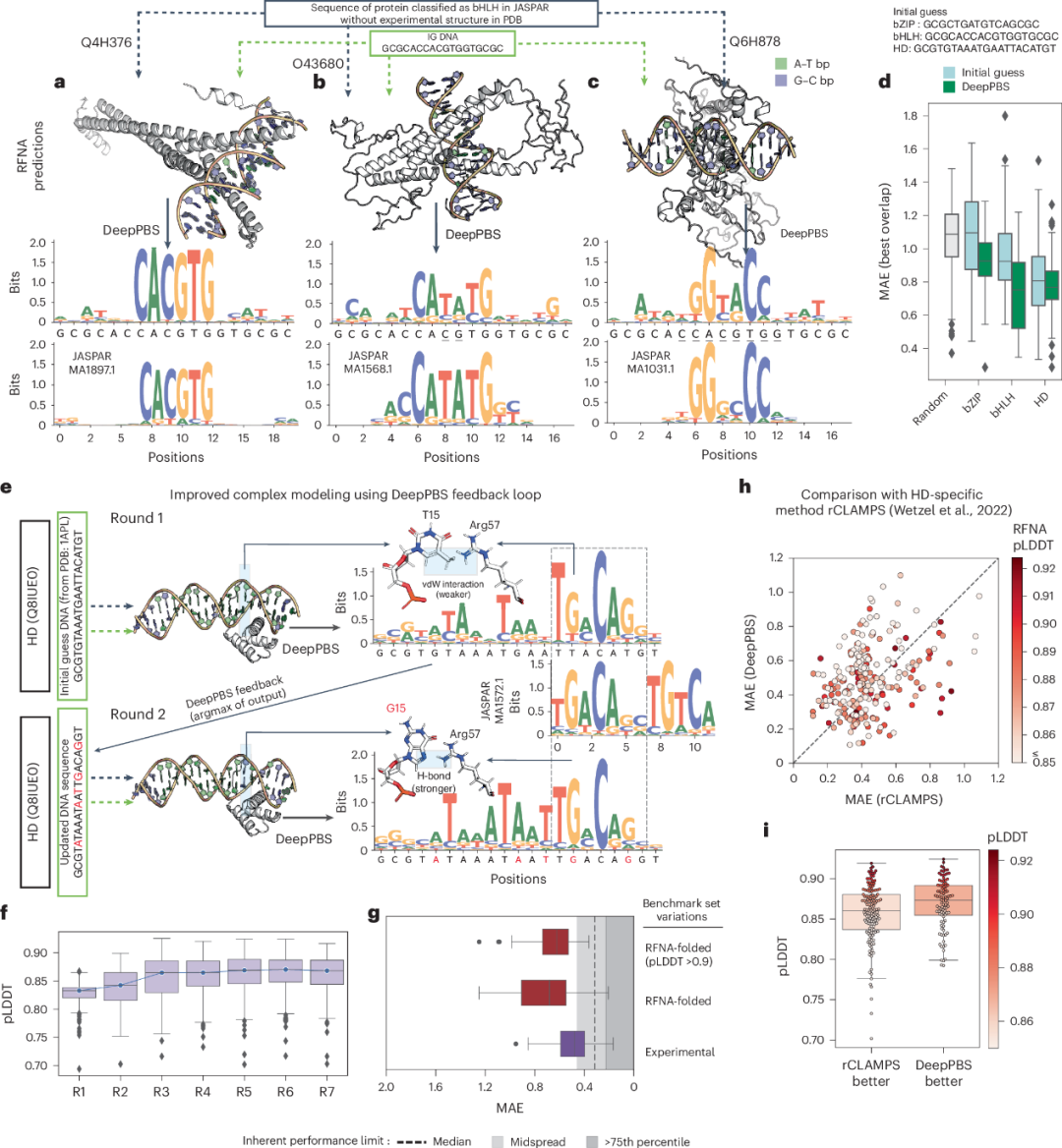
图 3
作者建议了一种在DeepPBS中处理预测结构的潜在方法。首先,根据相应的蛋白质家族,为每个感兴趣的蛋白质初步猜测其结合的DNA(IG DNA)序列。然后,使用RFNA预测蛋白质-DNA复合物的结构,接着用DeepPBS预测结合特异性。作者展示了这一过程(图3a–c),涉及在JASPAR中分类为碱性螺旋-环-螺旋(bHLH)的三种蛋白质。在这三种情况下,PDB中都缺乏实验蛋白质-DNA复合物结构。IG DNA(补充部分8)的中心具有增强子盒基序(“CACGTG”),已知这是bHLH家族的靶标。第一个例子(UniProt Q4H376;图3a)是Max同二聚体,DeepPBS预测的特异性与IG DNA的特异性非常接近。第二个例子(TCF21二聚体,O43680)更为复杂;IG DNA中的中央“CACGTG”基序假设错误,但DeepPBS成功预测了正确的基序为“CATATG”(图3b)。第三个例子(图3c,蛋白质OJ1581_H09.2,Q6H878)不符合任何增强子盒基序。然而,DeepPBS预测的结合特异性与实验数据非常接近(图3c)。
作者运行了DeepPBS流程,针对全长UniProt蛋白质序列(每个序列在JASPAR中都有唯一条目且没有复合物的实验结构),涵盖了三个不同的家族:bZIP、bHLH和HD家族。基于RFNA预测结构的DeepPBS预测表现出改进的MAE(即更接近实验数据),相比于IG DNA基线(图3d)。补充图9b展示了DeepPBS应用于MELD-DNA预测的小鼠CREB1蛋白复合物的实例。因此,DeepPBS可以从次优的DNA序列中获取预测结构,并预测出接近实验数据的结合特异性。
接下来,作者探索了是否可以将DeepPBS的预测作为反馈(循环)来增强蛋白质复合物的建模(并进一步改进DeepPBS的预测)。作者在图3e中展示了这个过程,使用人类TGIF2LY蛋白质(UniProt ID Q8IUE0,去除了无结构区域;补充部分8)作为示例。在第1轮中,作者将RFNA应用于该蛋白质序列和HD家族的IG DNA序列,并将预测的复合物作为DeepPBS的输入。对于IG DNA中的T15位点(图3e,第1轮),DeepPBS预测了对G的强烈偏好。在第1轮RFNA的输出中,Arg57和T15之间形成了一个氢键和一个范德华相互作用。这些相互作用理论上比G与Arg57之间可能形成的双齿氢键要弱。在第2轮中,作者通过选择DeepPBS输出的argmax(最优序列)来调整RFNA的输入(图3e,第2轮)。随后折叠的结构显示出G15和Arg57之间更强的双齿氢键相互作用,DeepPBS的预测结果也更接近实验数据(注意第2轮中A18、G19和T14的位置,对应于MA1572.1中的4-6位点;图3e)。
作者对一组HD单体序列重复进行了七轮DeepPBS预测过程。RFNA预测的置信度指标(预测的局部距离差异测试(pLDDT),LDDT反映了预测结构与复合物参考结构的相似性;在这些轮次中有所改善(图3f)。为了独立评估结构质量,作者计算了分子力学和泊松-玻尔兹曼表面积的结合能。从第1轮到第3+轮,稳定结构的数量(结合能<0 kJ mol−1)增加了,而它们的结合能分布向更低的值偏移。DeepPBS的表现在五轮中逐步提升。作者还通过RFNA重新折叠了基准集的数据点,并比较了(对于可处理的完整集(n = 98)和高置信度集,pLDDT >0.9,n = 31)与实验结构获得的相应性能(图3g)。性能有所下降,作者预计随着未来结构预测模型的改进,性能会有所提升。
DeepPBS预测结合特异性的方式与现有方法根本不同,后者仅基于蛋白质序列信息预测结合特异性。因此,无法与那些仅依赖蛋白质序列操作的现有家族特异性方法进行比较。然而,结合复合物结构预测方法,作者可以仅从蛋白质序列信息出发,使用DeepPBS预测结合特异性。这个过程可以与最近的HD家族特异性方法rCLAMPS进行比较。rCLAMPS可以预测HD蛋白质单体的核心6-mer结合特异性。性能的综合概览如图3h所示。在不同的数据部分,DeepPBS和rCLAMPS各有优劣。DeepPBS在pLDDT分数较高的情况下优于rCLAMPS(图3i)。因此,DeepPBS流程与rCLAMPS具有可比性,同时在家族和生物组装体中具有更广泛的适用性,并且不限于预测DNA核心结合区域。
参考资料
Mitra, R., Li, J., Sagendorf, J. M., Jiang, Y., Cohen, A. S., Chiu, T. P., ... & Rohs, R. (2024). Geometric deep learning of protein–DNA binding specificity.Nature Methods, 1-10.
原文地址:https://zhuanlan.zhihu.com/p/720483850 |
|

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 






 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 发表于 2024-11-8 11:19
发表于 2024-11-8 11:19



