金桔
金币
威望
贡献
回帖0
精华
在线时间 小时
|
声明:未经许可,禁止以任何形式转载。
本文目的
美国和欧盟,不仅拥有国际领先水准、行业占比极高的医疗器械制造企业,也是医疗器械消费大国\区域,均已建立相对完善的医疗器械法规体系,也都对全球医疗器械市场有着重要影响。
本期文章研究比较美国与欧盟医疗器械法规,直观对比得出两者在监管思路与方式方面的异同,对于需同时销往欧美两大市场的企业,无疑是一次让自身更加从容应对审核的知识充电。
1. 上市前要求
1.1 FDA将注册资料审查和体系考核分离,先开展文件资料的审核,审核通过后,在上市后进行抽查,以决定对某个厂家进行体系审核。
1.2 CE的Ⅰ类产品(非测量、非灭菌、非重复使用的产品)可使用自我声明的方式获得 MDR CE证书。其他类别通过公告机构审核后,则由公告机构颁发MDR证书。
1.3 美国:通过审核后(例如通过510(K)路径上市的产品),企业将获得官方发出的实质等同信函(SE Letter),并不会如同CE那样获得证书。重点:FDA 并不会发任何形式证书,市面上说FDA会有证书的均为咨询机构发的证书,是假证 请辨别!!
2. 认证周期
2.1 一般而言,整个CE认证周期并不明确,具体周期取决于医疗器械的类型和风险等级、技术文件的完整性、准备工作的充分程度等因素。
2.2 FDA注册,Ⅰ类产品在企业注册和列名完成后90天左右,将收到官方发出的注册编号。
Ⅱ类产品在递交510(K)文件后、FDA正式审核后、无法补的情况下,一次性通过审核所需时间为90天,而通常FDA给出的法补时间是180天。
3. 审核费用
3.1 CE认证MDR法规的费用包括:技术文件审核、现场审核、风险评估、申请机构和监管机构的认证费用和税费等环节收费。
具体费用还包括其他可能产生的费用如:质量体系的建立和维护费用、设备和测试工具的采购和维护费用、培训和人员费用等。
总体而言,CE认证MDR法规的费用并不明确,具体收费需根据产品类型、复杂程度、风险等级、技术文件的完整性、审核机构的不同等因素确定。
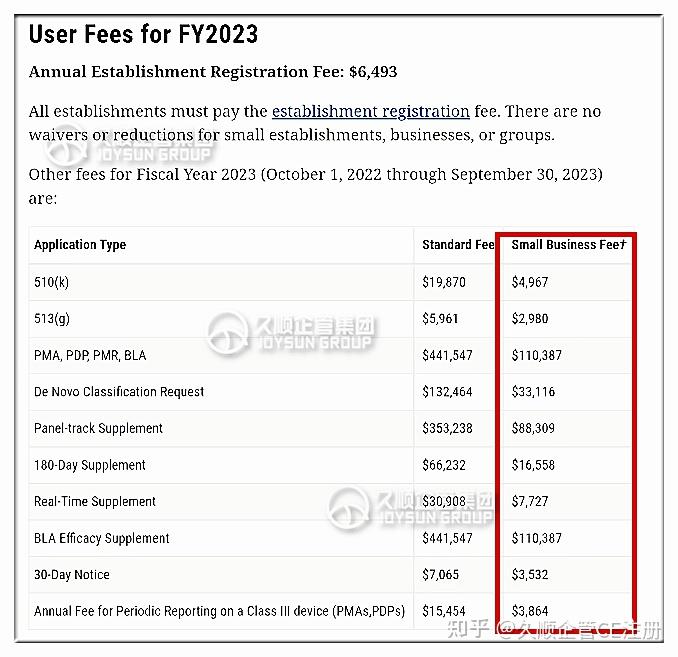
3.2 美国FDA在每个财政年度于官网发布下一年度的医疗器械企业年费和各项服务费的收费标准,费用由官方明确规定。
而且,满足一定条件企业即可申请小微企业的认定,当其被认定为小微企业后,510(k)的审核费用将获得较大力度的折扣优惠,相当于标准价的三分之一。
2023财政年度FDA的企业注册年费是$6493,510(k)标准收费是$19870,小微企业的此项收费则为$4967。
 |
|

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 






 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 发表于 2024-11-6 22:53
发表于 2024-11-6 22:53






 发表于 2024-11-6 22:55
发表于 2024-11-6 22:55



