用户名
UID
Email
密码
记住
立即注册
找回密码
只需一步,快速开始
微信扫一扫,快速登录
开启辅助访问
快捷导航
门户
Portal
社区
BBS
资讯
会议
市场
产品
问答
数据
专题
帮助
签到
每日签到
企业联盟
人才基地
独立实验室
产业园区
投资机构
检验科
招标动态
供给发布
同行交流
悬赏任务
共享资源
VIP资源
百科词条
互动话题
导读
动态
广播
淘贴
法规政策
市场营销
创业投资
会议信息
企业新闻
新品介绍
体系交流
注册交流
临床交流
同行交流
技术杂谈
检验杂谈
今日桔说
共享资源
VIP专区
企业联盟
投资机构
产业园区
业务合作
投稿通道
升级会员
联系我们
搜索
搜索
本版
文章
帖子
用户
小桔灯网
»
社区
›
C、IVD技术区
›
侧向层析技术
›
柱层析(过柱子),一文从入门到精通
图文播报
2026庆【网站十三周
2025庆【网站十二周
2024庆中秋、迎国庆
2024庆【网站十一周
2023庆【网站十周年
2022庆【网站九周年
返回列表
查看:
13955
|
回复:
0
[分享]
柱层析(过柱子),一文从入门到精通
[复制链接]
good
good
当前离线
金桔
金币
威望
贡献
回帖
0
精华
在线时间
小时
发表于 2024-10-25 20:56
|
显示全部楼层
|
阅读模式
登陆有奖并可浏览互动!
您需要
登录
才可以下载或查看,没有账号?
立即注册
×
我们常说过柱子其实就是柱层析分离,也叫柱色谱,1903年由俄国科学家首次发明使用,其把植物色素的溶液经过粉碎的碳酸钙粉末,发现从上到下在碳酸钙粉末上有不同的色带,通过对不同色带的分开提取,得到了较纯叶绿素,叶黄素等。
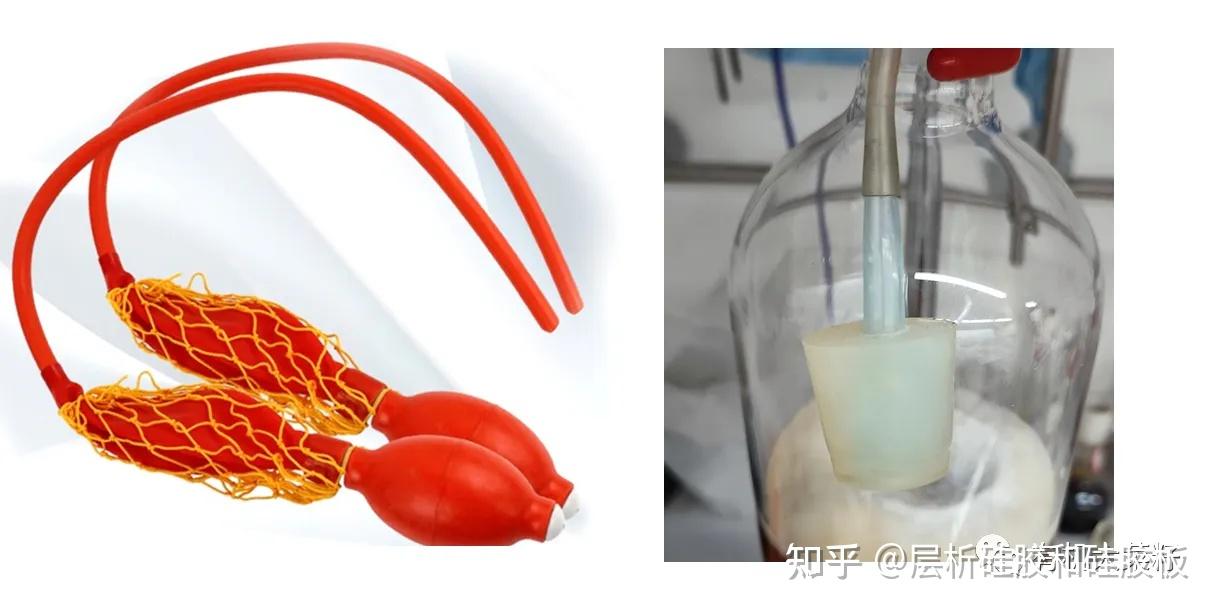
该方法经过经过百年的发展至今已经比较成熟,固定相也从单一的碳酸钙粉末发展到了氧化铝(又分为酸性、中性、碱性),硅胶,活性炭等,其中各大实验室最常用的便是硅胶,一般分为正相硅胶和反相硅胶!固定相(硅胶)一般要经过纯化和活性处理,颗粒大小应当均匀统一,理论上其颗粒越小、同单位质量表面积越大,吸附能力就越高,分离效果也就越好。但是实际使用中固定相的颗粒太小时往往会存在两个情况:一个是其质更轻,容易飘散在空气中(硅胶进入人体无法降解,越细越毒);二是颗粒小也会导致颗粒之间缝隙小,从而使溶剂的流速就太慢,耗时;以硅胶为例,目前已经商业化用量最大的硅胶尺寸为200-300目。过柱手段这块,除了前面说的正向循环制备,高效液相制备,过柱机这些自动分析的设备,手动过柱方面主要分为常压过柱,减压过柱,加压过柱三种。其中:减压过柱,因为洗脱剂流速过快,会损失大半部分塔板数,分离效果较差,适合分离TLC板子分的比较开的样品,尤其适合产物只有一个大极性杂质的样品;所有其大多数时间被用于化合物的预处理,过掉部分杂质后再仔细纯化。相关内容见:快速纯化样品的关键技术——硅胶短柱常压过柱是三者中分离效果最好的一种过柱方式,但是因为很多时候溶剂走的太慢,分离效果再好也会影响效率,所以这时候一般会给与压力,这就是所谓加压过柱;加压过柱压力不宜过高,一般手动的用双链球,自动的就用个养鱼泵,两者都可以如图改装一下。
前面的内容算一些介绍,下面将以正相柱层析为例子,我们讲一下常规的上样、过柱流程:
1,准备工作要做好选取柱子时,观察有无砂板,没有砂板的柱子记得底部填一些棉花,棉花不宜太厚也不宜太薄,能挡住硅胶不下来就好。这里面涉及到一个比较争议的问题,即甲醇过柱子会不会溶解硅胶?个人的经验是不会,一个解释见:是谁在鼓吹用甲醇过柱子会溶解硅胶?配套的准备好最小极性的的洗脱剂,紫外灯(实验室个人推荐手提式,随时可以照一照),加压设备,以及试管架!
2.选取合适的柱子,加入硅胶,浸润硅胶,拍平柱子的选取非常重要,大小应该根据产物的量来计算,一般可以分为:几毫克的柱子(全合成最终的选择);几十毫克的柱子(方法学扩底物);500毫克到1克的柱子(方法学做原料);5~100g左右的柱子,以及更大规模。
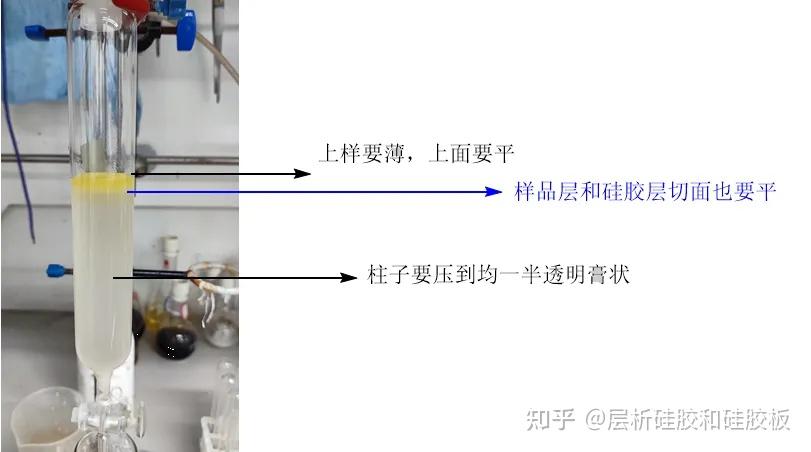
实验室最常见柱子直径和高度比值一般在1:2.5到1:15之间,上样时硅胶量是样品量的20~40倍左右。选取柱子的标准要实际根据你样品的TLC爬板情况来,如果杂质很多挨得很近,那么尽可能选大一丢丢的柱子,使你的样品正好能铺一小薄层(建议厚度在5mm以下),如果杂质很远,这个厚度自然也可以提高,但也不建议你样品厚度超过2cm,除非这个产物就是简单冲一冲就行。很多人会犯一个错误,就是想着我硅胶多加点,加的高高的,我样品上厚一点是不是也可以分离的很好?这个答案是否定的!理由是底下的样品和上面的样品不是在同一起跑线,这样只能的到一部分纯的,大部分是交叉的,过不纯不说,浪费时间浪费溶剂!这也是为什么样品要求铺薄的原因!
选取柱子后,加入硅胶时可以通过加料漏斗,同时一定要戴上口罩在通风橱里操作,原因是硅胶吸入人体后无法代谢,会将你的肺部打成筛子。硅胶粉是最毒的粉末之一,矽肺病约占全国尘肺病的50%!至于浸润硅胶,推荐的的流程如下:
①先把装好硅胶的硅胶柱竖直放好,硅胶表面用手拍平,
②硅胶柱底部连接水泵,把硅胶抽实,然后从上面倒入极性最小的洗脱剂,到洗脱剂刚要漏下时,关闭截止阀,防止溶剂抽到水泵。
③在加压状态,用最小的展开剂冲七八个柱体积就可以把硅胶浸润地均一。
3、上样
上样分为湿法上样和干法上样。湿法上样,建议的做法是:用胶头吸管吸取样品,靠近硅胶最上面切面,缓慢均匀滴加样品,直到覆盖一层,然后停止加样,给点压力让油状物沉浸到硅胶层里;油状物沉浸到硅胶层里后重复这种操作多次,直至全部上完样!有些新同学可能好奇为什么这样操作,而不是一次性把湿法上样的油状物一次性上完呢?答案是这种操作可以减少样品层的有效高度。即使还没有加洗脱剂,但当油状物浸入硅胶的那一刻,柱层析就已经开始,后面的样品相当于溶剂在推动前面的样品在硅胶里吸附、解吸附。至于干法上样,除非万不得已菜籽是不推荐的,固体样品更推荐打浆或者结晶:歪门邪道,乙酸乙酯VS水,超声波震荡助力高沸油状物结晶分享一种实验室重结晶的方法等等。干法上样一般会带来两个方面的问题;①拌样的过程中因为要升温旋蒸,增加了样品和硅胶之间反应的可能性;②干法上样就意味着样品层里会引入吸附剂,增加了样品层高度或柱子的尺寸,需要更多的时间,更多的溶剂。
如果不得已选择了,拌产物的硅胶用量一般以1~2倍样品量为宜。其上样直接把吸附好的粉慢慢倒进柱子就好,一些要求和湿法类似。
4、加入无水硫酸钠或石英砂
为了避免在加洗脱剂时撞击到样品层,使其不均匀,变形,影响分离效率,一般会在样品层上面铺一层无水硫酸钠或者石英砂。
也有见到加棉花的,但是掏出来就有点费劲了。菜籽个人喜欢铺无水硫酸钠,因为它还有干燥的功能,对于二氯甲烷这种易挥发吸热产生冷凝水的溶剂比较友好!
至于高度多少,就看自己的手法了!
5,梯度洗脱,采样收集
在上样之后,个人喜欢先用小极性溶剂(基本不会让产物下去的溶剂)冲柱2-3个柱体积,确保上下均一,然后选比例洗脱。
至于过柱子的洗脱剂怎么选?“洗脱剂的比例是你产物TLC爬板爬到Rf值约0.2时的比例。”这是菜籽看到很多书中以及很多博主分享的过柱经验,实际过程中也大差不差,个人来讲更喜欢缩小一倍梯度洗脱。
比如如果TLC爬板展开溶剂是PE/EA=2:1,菜籽更可能从PE/EA=4:1开始梯度洗脱,洗脱到PE/EA=2:1结束,一般效果更好。还有收集洗脱液的时候也是有技巧的,可以先选大一点的试管接收,TLC快到目标点了就换成小一点的试管,这样可以减少多接一点的可能性,不易包杂。
如果遇到杂质点和产物点挨得比较近的情况,特别是那些荧光又比较弱的,记得不要加压,应选常压过柱,每次接半管或更少就换管,也可以提高纯化几率。
6、柱子切忌不要及时处理!
很多新人有个习惯,过柱到了后期,TLC后面一两管没有产物,就默认柱子过完了,压干处理掉。等到一计算收率才发现收率偏低,或者去打谱发现谱图不正确,抓错点了,后悔莫及。所以菜籽建议您过完想要的点后柱子不要急忙处理,打核磁做收率都匹配之后再处理。如果忙,或者急着用这根柱子纯化其他化合物,建议换大极性良性混合溶剂冲一下,把这部分洗脱液单独留下来等鉴定结果。实际上,个人过柱的那些年,在看似出完产物的时候,都会换大极性溶剂冲一冲。遇到过有些东西一开始不了解形状,实际溶解性很差在柱子上断断续续出来,如用纯EA确定冲不出来了,但一旦换成二氯甲烷:甲醇体系,还会有!
原文地址:https://zhuanlan.zhihu.com/p/1006077805
回复
举报
返回列表
发表回复
高级模式
B
Color
Image
Link
Quote
Code
Smilies
您需要登录后才可以回帖
登录
|
立即注册
本版积分规则
发表回复
回帖后跳转到最后一页
关闭
官方推荐
/3
AI助手<小桔子>来了!
欢迎来交流,可以回答IVD行业各类问题!
查看 »
IVD业界薪资调查(月薪/税前)
长期活动,投票后可见结果!看看咱们这个行业个人的前景如何。请热爱行业的桔友们积极参与!
查看 »
小桔灯网视频号开通了!
扫描二维码,关注视频号!
查看 »
返回顶部
快速回复
返回列表
客服中心
搜索
洽谈合作
关注微信
微信扫一扫关注本站公众号
个人中心
个人中心
登录或注册
业务合作
-
投稿通道
-
友链申请
-
手机版
-
联系我们
-
免责声明
-
返回首页
Copyright © 2008-2024
小桔灯网
(https://www.iivd.net) 版权所有 All Rights Reserved.
免责声明: 本网不承担任何由内容提供商提供的信息所引起的争议和法律责任。
Powered by
Discuz!
X5.0 技术支持:
宇翼科技
浙ICP备18026348号-2
浙公网安备33010802005999号
快速回复
返回顶部
返回列表

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 






 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 发表于 2024-10-25 20:56
发表于 2024-10-25 20:56