金桔
金币
威望
贡献
回帖0
精华
在线时间 小时
|
登陆有奖并可浏览互动!
您需要 登录 才可以下载或查看,没有账号?立即注册


×
作者/编辑:黄潮勇——北京理工大学生命学院博士生 (微信公众号: My BioWorld)
导读:说起基因编辑领域的华人科学家,除了张锋以外,我首先能想到的就是David Liu,而在他众多的天才发明中,我最膜拜的就是碱基编辑器了。如果说CRISPR是基因编辑的皇冠,那么碱基编辑器就是皇冠上的明珠。2017年,David Liu入选“Nature年度十大人物”,而他开发的单碱基编辑技术也被评为“Science年度十大突破”。那么今天,我就给大家讲讲David Liu的碱基编辑器。
<hr/>如何精准、高效地对基因组进行修饰是生命科学领域研究的重要目标,而CRISPR/Cas9介导的基因编辑技术成为实现该目标的最强工具。传统的CRISPR/Cas9技术通过在靶点处产生DNA双链断裂(DSB),从而诱发细胞内的同源重组(HR)和非同源末端连接(NHEJ)修复途径,进而实现对基因组DNA的定点敲除、替换、插入等修饰。然而,DSB引发的DNA修复很难实现高效稳定的单碱基突变。
单核苷酸变异会导致大约2/3人类遗传病的发生,也是许多动植物重要性状变异的遗传基础,因此开发一种精准且能够高效实现单碱基替换的技术尤为重要,David Liu实验室开发的碱基编辑器就是为此而生的。David Liu实验室开发了三种不同的碱基编辑器,分别是胞嘧啶碱基编辑器(CBE)、腺嘌呤碱基编辑器(ABE)和先导编辑器(Prime Editor),这些碱基编辑器在工作时不依赖DSB的产生,也不需要供体DNA的参与。
那么接下来就让我来简单介绍一下这三种碱基编辑器
<hr/>1、胞嘧啶碱基编辑器
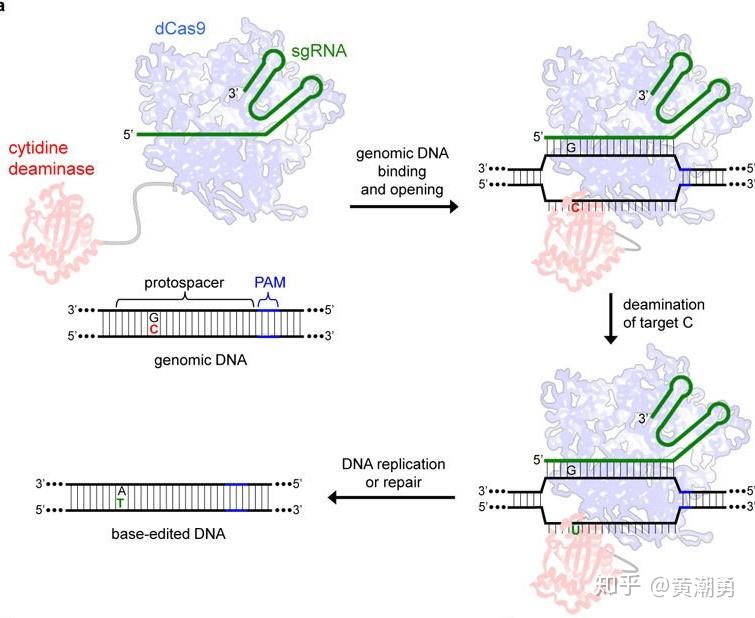
【图1】CBE的工作原理
CBE的核心组成元件是nCas9或dCas9和胞嘧啶脱氨酶,Cas9蛋白与胞嘧啶脱氨酶组成融合蛋白 [1]。具体工作原理:当融合蛋白在sgRNA的引导下靶向基因组DNA时,胞嘧啶脱氨酶可结合到由Cas9蛋白、sgRNA及基因组DNA形成的R-loop区的ssDNA处,将该ssDNA上一定范围内的胞嘧啶(C)脱氨变成尿嘧啶(U),进而通过DNA复制或修复将U转变为胸腺嘧啶(T),最终实现C•G碱基对至T•A碱基对的直接替换。
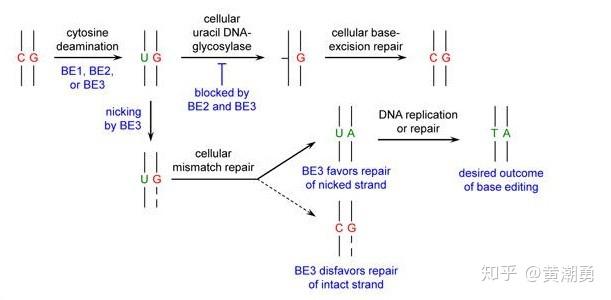
【图2】CBE的升级
第一代胞嘧啶碱基编辑器:rAPOBEC1-XTEN-dCas9
第一代胞嘧啶碱基编辑器BE1由rAPOBEC1(大鼠胞嘧啶脱氨酶)和完全失去切割活性的dCas9组成 [1],可以在体外实现有效的碱基编辑(25%~40%),但在哺乳动物细胞内的编辑效率大大下降(0.8%~7.7%)。主要原因是细胞内存在尿嘧啶DNA糖基化酶(UDG),UDG可识别U•G错配,并切割尿嘧啶和磷酸骨架之间的糖苷键,通过细胞内的碱基切除修复途径(BER)将U逆转为C。
第二代胞嘧啶碱基编辑器:rAPOBEC1-XTEN-dCas9-UGI
为了抑制UDG的作用,David Liu实验室在BE1的基础上融合了来自噬菌体PBS的尿嘧啶DNA糖基化酶抑制剂(UGI),开发了第二代胞嘧啶碱基编辑器BE2 [1]。UGI能够抑制人类和细菌中UDG的作用,提高编辑效率,经测试BE2的编辑效率比BE1提高了3倍。
第三代胞嘧啶碱基编辑器:rAPOBEC1-XTEN-nCas9-UGI
为了进一步提高编辑效率,David Liu实验室结合细胞的内源修复机制开发了第三代碱基编辑器BE3 [1]。BE3将BE2中的dCas9替换为nCas9(D10A),可特异性地在非编辑链上产生一个缺口,进而刺激细胞内的碱基错配修复途径(MMR),以含有U的编辑链作为模板进行修复,从而增加编辑效率。BE3的编辑效率比BE2提高了2~6倍,平均约为37%,该碱基编辑器是目前较为广泛使用的CBE版本。
第三代胞嘧啶碱基编辑器的不同子版本
BE3编辑的活性窗口可覆盖sgRNA的第4~8位,依据不同的研究目的需要对编辑活性窗口做相应的改变。当需要精准地改变某个特定的碱基C时,过大的活性窗口会导致窗口内非靶标碱基C的编辑。David Liu实验室在BE3的基础上,通过突变胞嘧啶脱氨酶上与DNA作用的关键活性位点,开发了YE1-BE3、YE2-BE3、EE-BE3和YEE-BE3,从而将编辑活性窗口由5nt缩小至1~2nt,但同时对靶标C的编辑效率有所降低 [2]。
第四代胞嘧啶碱基编辑器:rAPOBEC1-XTEN-nCas9-2UGI
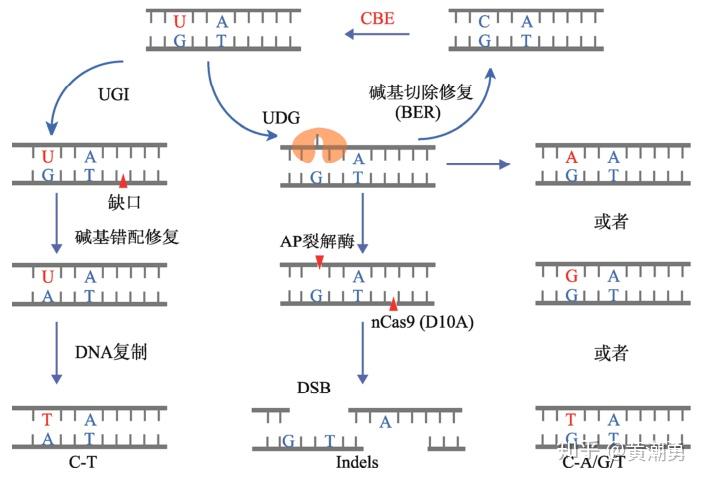
【图3】BE3编辑产生不同的产物
BE3编辑后依旧会产生不同的产物,主要表现为两个方面: (1) BE3除了将C转换为T之外,也有一定几率将C转换为G或A [1]。这是由于UDG可将U切除形成无嘧啶位点(AP),经过跨损伤合成(TLS)聚合酶作用及DNA复制等,也有一定几率将C转换为其他碱基。(2) 在编辑过程中会产生少量的Indels [1],这是由于形成的AP位点会在AP裂解酶或自发裂解下产生一个缺口,进而与nCas9在非编辑链产生的缺口刚好形成一个DSB,经过NHEJ修复途径后产生Indels产物。
为了减少不必要的编辑产物,David Liu实验室在BE3的基础上融合了第二个拷贝的UGI,增强对UDG的抑制作用,构建了第四代胞嘧啶碱基编辑器BE4 [3]。与BE3相比,BE4不仅提高了碱基编辑效率,并且C至A或G的转换频率降低了2.3倍。
第四代胞嘧啶碱基编辑器的不同子版本
为了降低Indels的发生,David Liu实验室在BE4的基础上融合了来源于噬菌体Mu的Gam蛋白,构建了BE4-Gam [3]。Gam蛋白可结合于DSB的末端防止其降解,进而阻止了NHEJ修复途径的发生。
细胞内胞嘧啶碱基编辑器的表达水平是影响其编辑效率的重要因素,David Liu实验室在BE4的基础上通过增加不同数量的核定位信号(NLS)以及使用不同公司优化的密码子序列等方法构建了BE4max和AncBE4max,这两种胞嘧啶碱基编辑器可在各种哺乳动物细胞内进行高效的编辑。
<hr/>2、腺嘌呤碱基编辑器
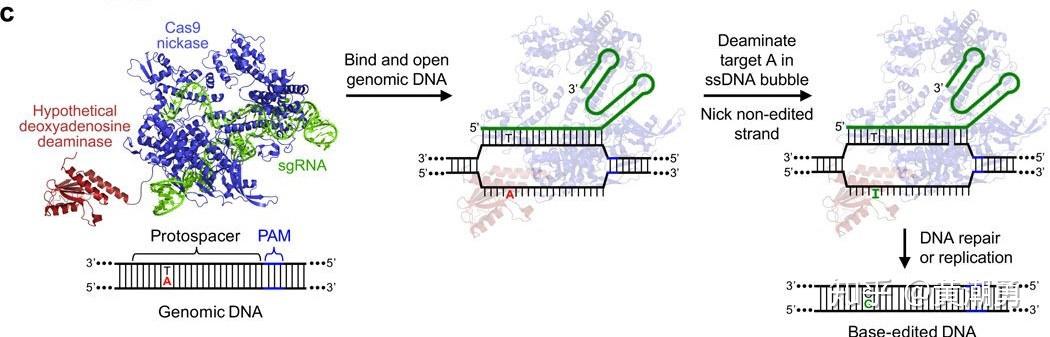
【图4】ABE的工作原理
ABE的核心组成元件是nCas9(D10A)和人工定向进化的腺嘌呤脱氨酶,Cas9与腺嘌呤脱氨酶组成融合蛋白 [5]。ABE的作用原理与CBE类似,即当融合蛋白在sgRNA的引导下靶向基因组DNA时,腺嘌呤脱氨酶可结合到ssDNA上,将一定范围内的腺嘌呤(A)脱氨变成肌苷(I),I在DNA水平会被当做G进行读码与复制,最终实现A•T碱基对至G•C碱基对的直接替换。ABE的开发打破了CBE仅能编辑C或G的限制,为碱基之间的相互转变提供了更多的可能性。相比CBE,ABE不需要抑制烷基腺嘌呤DNA糖基化酶(AAG)的活性。
腺嘌呤脱氨酶的改造
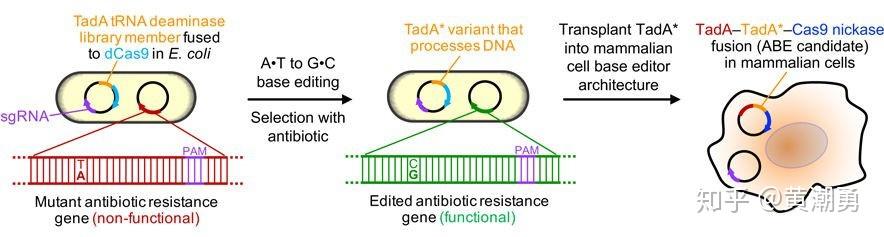
【图5】定向进化改造腺嘌呤脱氨酶
由于目前已知的腺嘌呤脱氨酶不能以DNA为底物对碱基A进行脱氨,因此必须对现有的腺嘌呤脱氨酶进行定向改造,这导致ABE的开发比CBE更具有挑战性。David Liu实验室选取大肠杆菌的腺嘌呤脱氨酶TadA为改造对象,将随机突变的TadA融合dCas9构建随机突变库,通过ABE恢复氯霉素、卡那霉素、壮观霉素等抗性基因的功能,结合易错PCR、DNA重排等定向进化策略,进行相应的抗生素筛选,先后经过7轮进化与改造后,成功筛选到了能直接作用于ssDNA的腺嘌呤脱氨酶,在人类细胞中建立了目前效率最高且应用最广的ABE版本ABE7.10(ecTadA-ecTadA*-nCas9) [5]。
ABE7.10将nCas9与野生型腺嘌呤脱氨酶ecTadA和经过定向进化的腺嘌呤脱氨酶ecTadA*二聚体融合,在人类细胞中编辑效率约为50%,编辑活性窗口可覆盖sgRNA的第4~9位。
腺嘌呤碱基编辑器的升级
无论在哺乳动物还是植物等生物中,ABE7.10均可保证高精度的碱基替换和较少的Indels发生,因此对ABE7.10的优化主要表现在提高编辑效率、扩大编辑活性窗口和扩大编辑范围。
David Liu实验室在ABE7.10的基础上通过融合不同数量的NLS以及使用不同公司优化的密码子序列构建了ABEmax,提高了对碱基A的替换效率[6]。随后,David Liu实验室对ABEmax进行改造,构建了CP1012-ABEmax、CP1028-ABEmax、CP1041-ABEmax和CP1249-ABEmax,这些版本的编辑器在保证编辑效率与ABEmax相当的同时,在一定程度上将4~9位的编辑活性窗口拓展为4~12位[7]。
<hr/>3、先导编辑器
CBE和ABE组合使用可以有效地进行4种碱基转换(C→T, G→A, A→G, T→C),而无需产生DSB,然而除了这4种碱基转换,对另外8种碱基转换(C→A, C→G, G→C, G→T, A→C, A→T, T→A, T→G)以及碱基的插入和缺失,依然缺乏有效的研究工具。
为了解决这个问题,David Liu实验室开发出先导编辑器(Prime Editor, PE),PE在不依赖DSB和供体DNA的条件下便可有效实现所有12种碱基转换,此外还能有效实现多碱基的精准插入(最多可插入44bp)和删除(最多删除80bp) [8]。
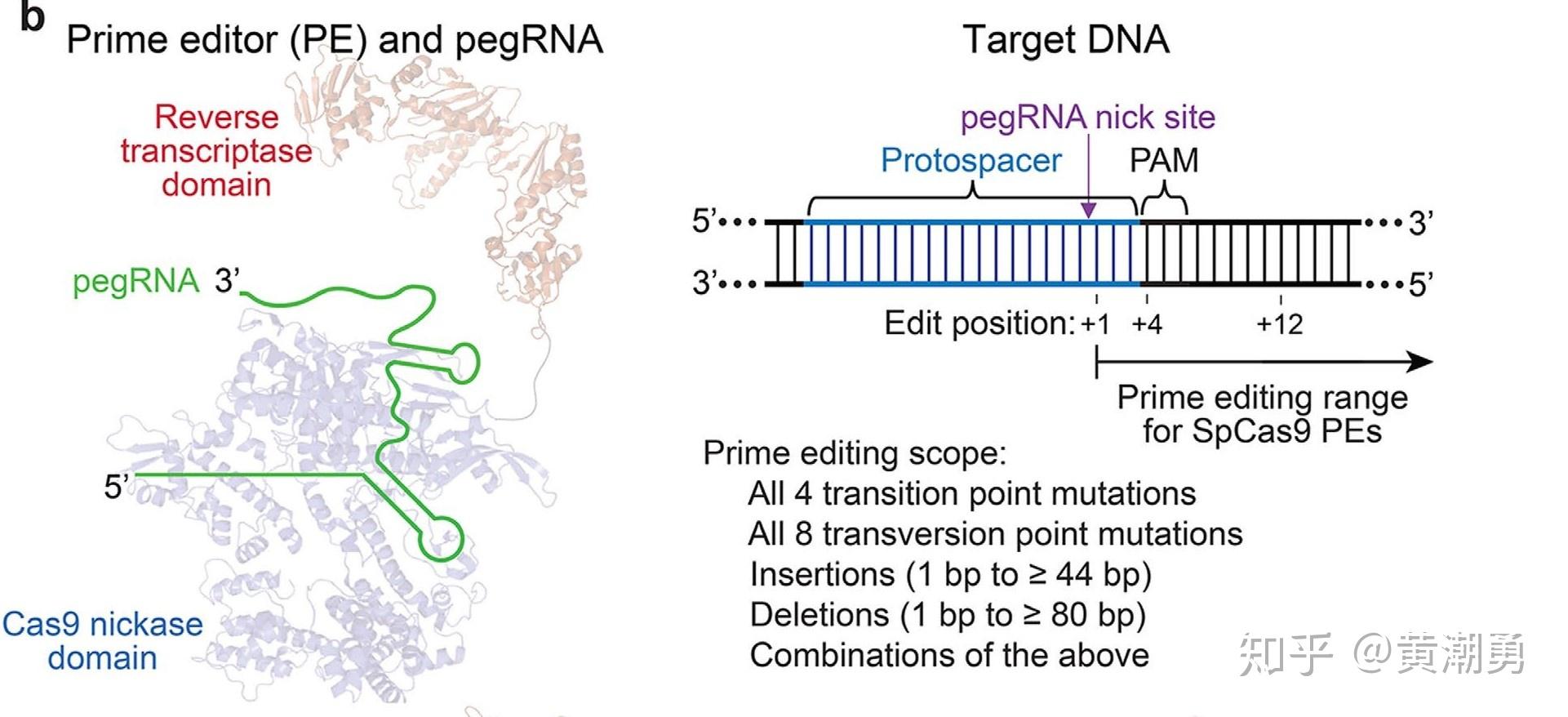
【图6】PE的组成
PE以CRISPR/Cas9系统为基础,在两方面加以改造。首先是改造sgRNA,在其3'末端增加一段RNA序列,获得的RNA被称作pegRNA;其次是将nCas9(H840A)与逆转录酶融合,获得新的融合蛋白 [8]。pegRNA上新增加的RNA序列有双重角色,一段序列作为引物结合位点(PBS),与断裂的靶DNA链3'末端互补以起始逆转录过程,另一段序列作为逆转录模板,携带了目标点突变或插入缺失突变以实现精准的基因编辑。
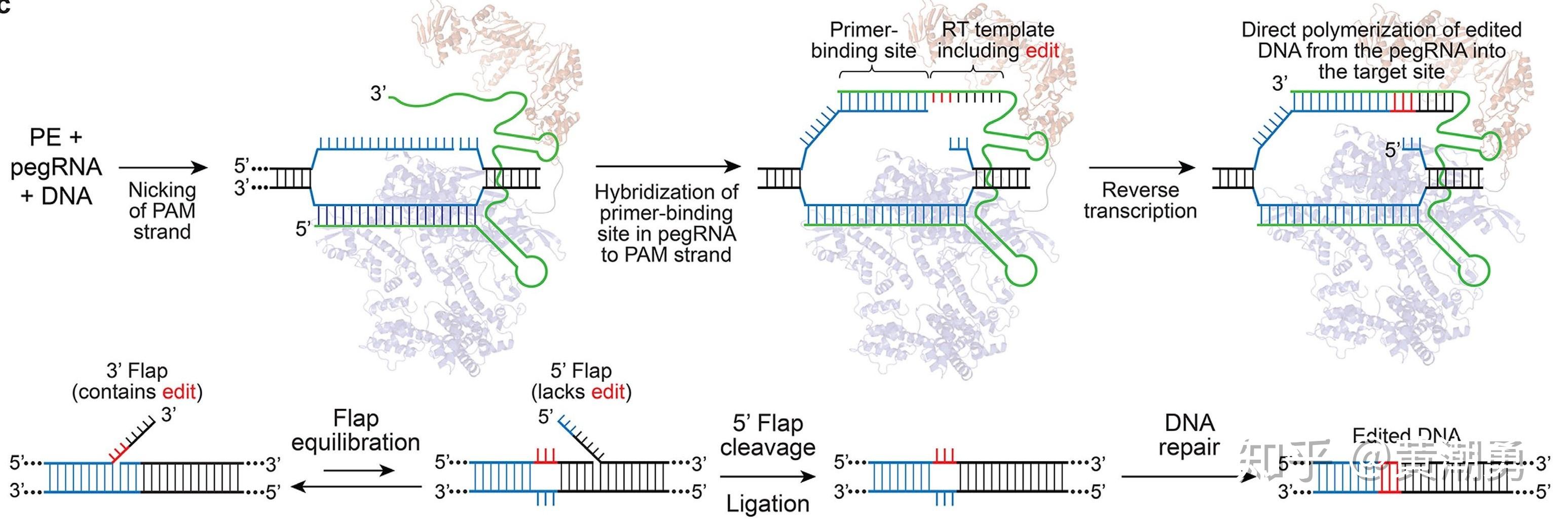
【图7】PE的工作原理
PE的工作原理如图7所示,首先是在pegRNA的引导下,nCas9切断含PAM的靶DNA链,断裂的靶DNA链与pegRNA的3’末端PBS序列互补并结合,之后逆转录酶发挥功能,沿逆转录模板序列开始逆转录反应。反应结束后DNA链的切口处会形成处在动态平衡中的5’flap和3’flap结构,其中3’flap结构的DNA链携带有目标突变,而5’flap结构的DNA链则无任何突变。细胞内5’flap结构易被结构特异性内切酶识别并切除,之后经DNA连接和修复便实现了精准的基因编辑 [8]。
先导编辑器的升级
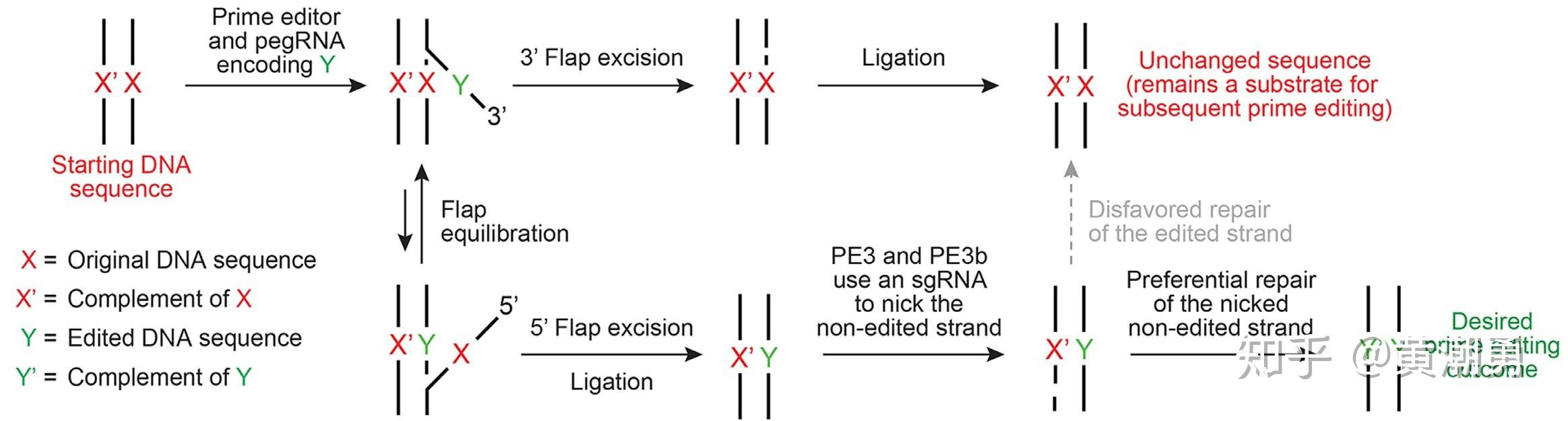
【图8】PE的升级
David Liu实验室首先将野生型的鼠白血病病毒(M-MLV)逆转录酶与nCas9融合,构建出第一代先导编辑器PE1。PE1在293T细胞中的点突变效率为0.7~5.5%, 碱基插入/删除的效率为4~17% [8]。随后他们通过优化M-MLV逆转录酶得到第二代先导编辑器PE2,其碱基突变和插入/删除的效率较PE1有两倍以上的提高 [8]。
PE1/2系统只编辑双链DNA的一条链,另一条非编辑链需进一步的DNA修复以完成精准编辑。理论上,通过nCas9切断非编辑链可以有效提高该链的修复效率,为此David Liu实验室在PE2的基础上,增加可切断非编辑链的sgRNA,获得PE3和PE3b []。PE3和PE3b的编辑效率较PE2提升了将近3倍,在293T细胞中的最高编辑效率可达78%。当然,由于使用了两条sgRNA,PE3和PE3b的引入Indels的风险也随之提高,这是PE3未来需要加以改进的不足之处。
Nature杂志评论这一技术是“超精确的新型基因编辑工具”;Science杂志评论它是“超越CRISPR”的重大突破”;哈佛大学教授,CRISPR先驱George Church盛赞这一成果“朝着正确方向迈出的一大步”。
<hr/>结语:像David Liu这种大神级别的人物,我除了膜拜还是膜拜,如果你去查他们实验室这几年发的文章,你就会发现,他们发CNS及其子刊的频率是以天或周来计算的,而我们发SCI也只能用月甚至年来计算。我对David Liu的编辑编辑器是很钟爱的,很多文章我都仔细看了,也一直想在实验室建立这一套系统,然而我发现每次我还没来得及设计实验方案,他们的系统又升级了!对于这一点,我真的是无Fuck说!
参考文献
[1] Nature(2016) 533:420-424
[2] Nat Biotechnol(2017) 35:371-376
[3] Sci Adv(2017) 3:eaao4774
[4] Nat Biotechnol(2018) 36:843-846
[5] Nature(2017) 551:464-471
[6] Nat Biotechnol(2018) 36:843-846
[7] Nat Biotechnol(2019) 37:626-631
[8] Nat Biotechnol(2020) 38:620-628
原文地址:https://zhuanlan.zhihu.com/p/141987848 |
|

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 






 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 发表于 2024-10-5 07:43
发表于 2024-10-5 07:43



