金桔
金币
威望
贡献
回帖0
精华
在线时间 小时
|
登陆有奖并可浏览互动!
您需要 登录 才可以下载或查看,没有账号?立即注册


×
基因测序技术
基因测序技术也称作DNA测序技术,即获得目的DNA片段碱基排列顺序的技术,获得目的DNA片段的序列是进一步进行分子生物学研究和基因改造的基础。
基因测序技术的发展历史
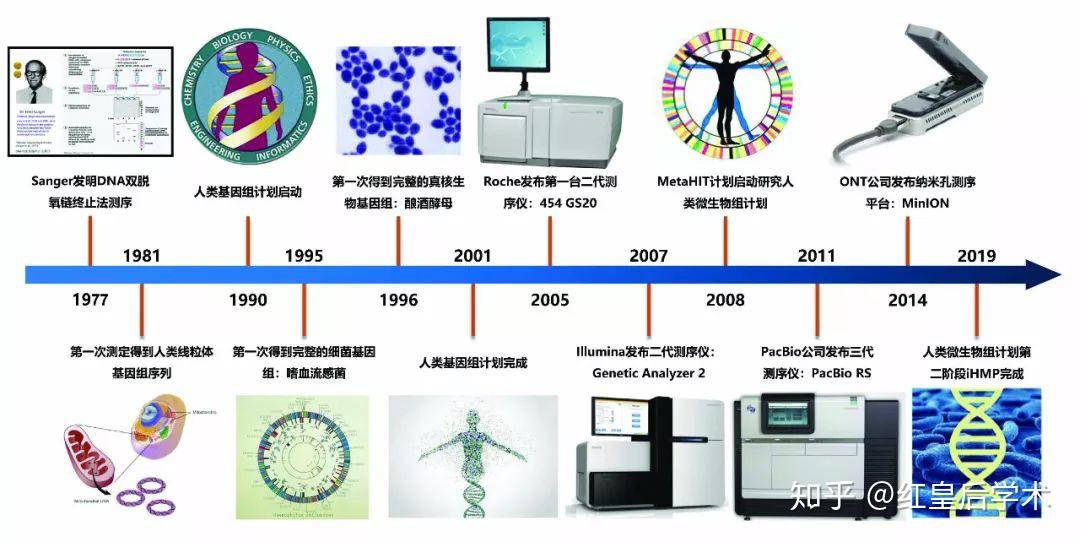
基因测序技术发展历史
1977年,Walter Gilbert和Frederick Sanger发明了第一台测序仪,并应用其测定了第一个基因组序列,噬菌体X174,全长5375个碱基。
由此开始,人类获得了探索生命遗传本质的能力,生命科学的研究进入了基因组学的时代,到至今为止的四十年时间内,测序技术已取得了相当大的发展,从第一代发展到了第三代测序技术。
Sanger所发明的测序方法被称为第一代测序技术,该技术直到现在依然被广泛使用,但是其一次只能获得一条长度在700~1000个碱基的序列,无法满足现代科学发展对生物基因序列获取的迫切需求。
高通量测序 (High-Throughput Sequencing, HTS) 是对传统Sanger测序的革命性变革,其解决了一代测序一次只能测定一条序列的限制,一次运行即可同时得到几十万到几百万条核酸分子的序列,因此也被称为新一代测序 (Next Generation Sequencing, NGS)或第二代测序。
第二代测序技术虽然测序的通量大大增加,但是其获得单条序列长度很短,想要得到准确的基因序列信息依赖于较高的测序覆盖度和准确的序列拼接技术,因此最终得到的结果中会存在一定的错误信息。
因此,科研人员又发明了第三代测序技术也称为单分子测序技术,该技术在保证测序通量的基础上,对单条长序列进行从头测序,能够直接得到长度在数万个碱基的核酸序列信息。
测序成本的变化
除了测序通量和读长的进步之外,测序技术的大范围应用最主要应该归功于成本的下降,在早期只有第一代测序技术之时,人类基因组计划耗资30亿美元才获得了大部分的人类基因组信息,这样高昂的成本显然不是常规科学研究者能够承受的。
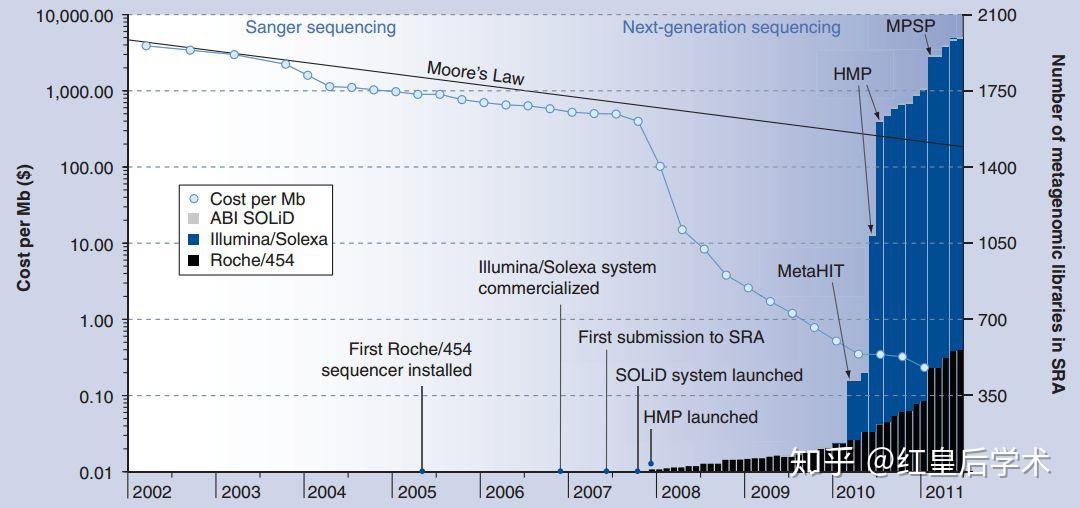
基因测序技术成本变化
新一代测序技术的发明和应用大大降低了获取核酸序列所需的成本,其打破了摩尔定律的限制,使得获得基因序列所需的金钱出现了断崖式的下降,在2008年,全基因组测序的成本降至20万美元,到2010年,该费用已经可以控制在10000美元以内,目前,测定一个人类的全基因组只需要不到1000美元即可完成。
测序技术的发展方向
目前,基因测序技术已经在众多领域得到广泛应用,包括生物的基因组图谱绘制、环境基因组学和微生物多样性、转录水平动态响应及其调控机制,疾病相关基因的确定和诊断、表观遗传学和考古学、物种进化演替过程等等。
就当前市场形势看来第二代短读长测序技术在全球测序市场上仍然占有着绝对的优势地位,但第三代测序技术的应用也已在近几年实验了快速发展。
未来基因测序技术发展方向:
- 更快的序列获取速度;
- 更准确的碱基识别方式;
- 更长的单条测序序列长度;
- 更轻便的测序仪器平台;
- 更简便的操作过程;
- 更便宜的测序价格。
<hr/>第一代测序技术
1975年由Frederick Sanger所提出的链终止法以及1977年由Walter Gibert所发明的链降解法被称为第一代测序技术。
1977年,Walter Gilbert和Frederick Sanger发明了第一台测序仪,并应用其测定了第一个基因组序列,噬菌体X174,全长5375个碱基。Walter Gilbert和Frederick Sanger也因在测序技术中的贡献获得了1980年诺贝尔化学奖。
技术原理
目前,基于第一代测序技术的测序仪几乎都是采用Sanger提出的链终止法。
链终止法测序的核心原理是ddNTP的2'和3'端都不含羟基,因此在合成核酸链的过程中无法形成磷酸二酯键,从而导致DNA合成反应中断。
在测定待测核酸片段的序列时,向反应体系中加入一定比例的带有放射性同位素标记的4种ddNTP,利用DNA聚合酶来延伸结合在待测核酸模板上的引物,直到掺入一种链终止核苷酸为止,最终会得到一组长度各相差一个碱基的链终止产物,这些产物可通过高分辨率变性凝胶电泳分离并根据其长度排序,凝胶处理后可用X-光胶片放射自显影进行检测,从而确定目的核酸片段各个位置的碱基。
完整的测序过程分为4步:
1. DNA碎片化:
如要利用Sanger测序方法进行完整基因组的测定,首先要将提取得到的样品完整DNA打碎,形成DNA片段,如只是测定单个目的基因的序列,则无需进行DNA碎片化。
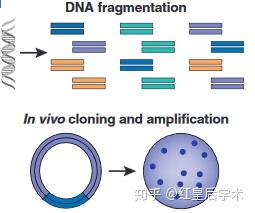
DNA碎片化和体外克隆
2. PCR扩增和体外克隆:
针对特定目的核酸片段的测序,首先要对目的测序区域进行PCR扩增;而针对碎片化DNA的测序,则要将碎片化的DNA片段通过克隆的方式连接到质粒载体中;对于部分PCR产物的测序也可以对其进行克隆,以保证测序样品的纯度和浓度。
3. ddNTP法循环测序:
向得到的待测样品中分别加入4种dNTP和4种ddNTP,从而得到不同位置匹配终止的序列。
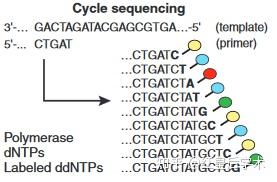
ddNTP循环测序
4. 凝胶电泳获得序列:
对得到的序列进行凝胶电泳,根据碱基的顺序和位置确定序列信息。
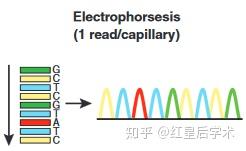
电泳确定序列
第一代测序技术的优势和劣势
优势:
- 第一代测序技术的准确性远高于二、三代测序,因此被称为测序行业的“金标准”;
- 第一代测序每个反应可以得到700-1000bp的序列,序列长度高于二代测序;
- 第一代测序价格低廉,设备运行时间短,适用于低通量的快速研究项目。
劣势:
- 第一代测序技术一个反应只能得到一条序列,因此测序通量很低;
- 第一代测序技术虽然单个反应价格低廉,但是获得大量序列的成本很高。
第一代测序技术的应用
- PCR产物测序:
对目的基因的PCR产物进行测序,得到目的基因序列;
- 重测序:
突变、SNPs、插入或缺失克隆产物的验证;
- 分型分析:
微生物和真菌分类学鉴定、HLA分型、病毒分型等;
- 临床应用:
肿瘤突变基因的检测和肿瘤个体化治疗,致病基因位点明确并且数量有限的单基因遗传病检测;
- 对新一代测序技术的结果进行验证。
第一代测序技术常见问题及解决方法
样品测序无信号
此时测序完全失败,最可能的原因是待测样品出现了降解或引物失效,从而导致测序引物与待测样品无法结合。
此时探索造成测序失败的具体原因并无实际意义,最快速、简便的办法是重新提供质量合格的引物和样品再次进行测序。
样品测序信号差
此种情况可能是引物或模板的质量不高或是引物和模板的匹配性不好引起的,但最有可能的原因是待测样品浓度偏低。
待测样品浓度偏低可能是由于PCR效率较低,也可能是PCR与测序间隔时间过长,导致PCR产物降解。建议PCR完成后尽快进行测序,如果PCR产物浓度本身较低,可以使用PCR产物作为模板进行二次PCR,也可以对PCR产物进行克隆后,再进行测序。
样品测序衰减
可能是由于待测样品包含特殊的核酸结构,如重复序列、回文结构、发卡结构、GC富集区、AT富集区等。
由于是样品本身结构问题,因此,无法通过优化测序反应解决,应从待测样品另一端进行反向测序,之后两端的测序结果拼接得到完整序列。
样品测序中断
此种情况是由于待测样品包含特殊高级结构,导致碱基无法与模板结合,DNA聚合酶无法继续延伸。
此情况与样品测序衰减解决办法相同,均为从待测样品另一端进行反向测序,经拼接后可以得到完整序列。
样品测序移码
测序从起始位置即发生移码是由于引物发生降解,应重新提供引物进行测序;
如测序过程中出现局部移码的现象,则可能是待测样品包含特殊高级结构,应当反向测序后拼接得到完整序列。
样品测序套峰
套峰细分的话有如下几种情形:
- 全双峰:
如样品为克隆后质粒,则质粒中含有多个引物结合位点;
如样品为PCR产物,则含有非特异性扩增。
- 前端双峰:
如样品为克隆后质粒,则其含有多个引物结合位点,并且其中一套模板出现测序中断的现象;
如样品为PCR产物,则PCR产物中含有多个引物结合位点,或者PCR产物中含有引物二聚体等小片段污染。
- 中间双峰:
如样品为克隆后质粒,则质粒并非单克隆;
如样品为PCR产物,则部分产物中具有碱基缺失现象,或目的基因为等位基因导致PCR产物自身不纯。
- 后端双峰:
如样品为克隆后质粒,则质粒并非单克隆;
如样品为PCR产物,则部分产物中具有碱基缺失现象。
解决办法:
- 针对二聚体及小片段干扰的情况,可以使用切胶回收的方法纯化PCR产物;
- 针对含有多个引物结合位点的情况,应当更换测序引物;
- 针对PCR产物出现碱基缺失的情况,可以使用克隆后测序以排除碱基缺失的产物;
- 针对非单克隆的情况,应在确认克隆无误的前提下重新挑取单克隆进行测序;
- 针对PCR产物含有非特异性扩增的情况,应优化PCR反应条件去除非特异性扩增,重新制备样品测序;
- 针对等位基因具有双模板的情况,应当采用克隆测序以保证单次测序样品序列一致。
样品测序底峰干扰
可能是由于测序引物不纯导致的,应当采用高纯度的引物 (PAGE级) 或重新提供引物进行测序。
<hr/>第二代测序技术
高通量测序技术 (High-throughput sequencing, HTS) 是对传统Sanger测序技术革命性的变革,可以一次对几十万到几百万条核酸分子进行序列测定,因此也称其为下一代测序技术 (Next Generation Sequencing, NGS),高通量测序技术的出现使得对一个物种的转录组和基因组进行细致全貌的分析成为可能。
技术平台
经过科研人员的不断开发和改进,目前成熟的第二代测序技术共有3种,分别为Roche公司的454技术、ABI公司的SOLiD技术和Illumina公司的Solexa技术。
Roche/454
该技术由Jonathan Rothberg于2005年发明,该技术是第一个被发明的二代测序技术,该技术引领生命科学的研究进入高通量测序时代。该技术的基本原理是:一个片段 = 一个磁珠 = 一条读长,DNA片段无需进行荧光标记,无需电泳,边合成变测序,碱基在加入到序列中时,会脱掉一个焦磷酸,通过检测焦磷酸识别碱基,因此也被称为焦磷酸测序。
ABI/SOLiD
SOLiD技术是由连接酶测序法发展而来,Lerroy Hood在上世纪80年代中期利用连接酶法设计了第一台自动荧光测序仪。SOLiD以四色荧光标记寡核苷酸的连续连接合成为基础,取代了传统的聚合酶连接反应,可对单拷贝DNA片段进行大规模扩增和高通量并行测序。
Illumina/Solexa
Illumina公司的第二代测序仪最早由Solexa公司研发,其同样为边合成边测序,该技术在测序的过程中,加入改造过的DNA聚合酶和带有4种荧光标记的dNTP,因为dNTP的3'羟基末端带有可化学切割的部分,它只容许每个循环掺入单个碱基,此时,用激光扫描反应板表面,根据dNTP所带的荧光读取每条模板序列每一轮反应所聚合上去的核苷酸种类,经过“合成-清洗-拍照”的循环过程,最终得到目的片段的碱基排列顺序。
技术原理
Roche/454
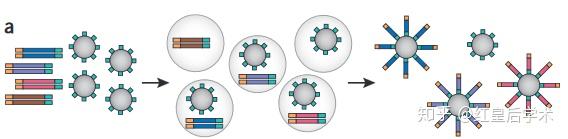
Roche/454技术原理
1. Preparation
454测序技术利用喷雾法将待测DNA打断成300-800bp长的小片段,并在片段两端加上不同的接头,或将待测DNA变性后用杂交引物进行PCR扩增,连接载体,构建单链DNA文库。
2. Emulsion PCR
在PCR反应前,将包含PCR所有反应成分的水溶液注入高速旋转的矿物油表面,形成被矿物油包裹的无数个小水滴,每一个小水滴即为一个独立的PCR反应空间,理想状态下,每一个小水滴只包含一个DNA模板和一个磁珠,磁珠表面含有与接头互补的DNA序列,经过PCR扩增后,磁珠上会富集大量序列相同的PCR产物,从而达到测序所需DNA量的要求。
3. Sequencing
测序时,需将磁珠固定在特制的PTP平板上。这种平板上含有许多直径约为44μm的小孔,每个小孔仅能容纳一个磁珠,通过这种方法来固定每个磁珠的位置。
启动测序反应后,每次向PTP平板中加入一种dNTP,如果能与待测序列配对,则会在碱基连接在模板上之后释放焦磷酸,焦磷酸通过ATP硫酸化学酶激活荧光素酶产生荧光,通过PTP板另一侧的CCD照相机记录荧光,从而确定目的模板的核酸序列。
ABI/SOLiD
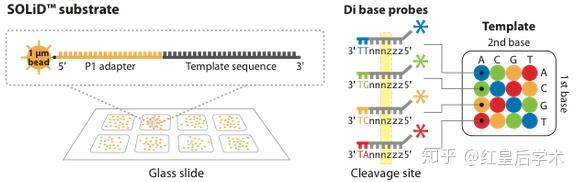
ABI/SOLiD技术原理
SOLiD测序技术与454技术的原理比较类似,同样是采用油包水的方式进行Emulsion PCR。
不同之处在于SOLiD形成的小水滴要比454系统小得多,只有1μm大小,并且在PCR扩增的同时对扩增产物的3'端进行修饰,为下一步的测序做准备。
在PCR完成之后,SOLiD技术进行测序时,其反应底物不是dNTP也不是ddNTP,而含有8个碱基的单链荧光探针混合物,在测序时,这些探针按照碱基互补规则与单链DNA模板链配对,不同的探针的5'末端分别标记不同颜色的荧光染料,每两个碱基确定一个荧光信号,相当于一次能决定两个碱基,因此,这种测序方法也被称为两碱基测序法。
Illumina/Solexa
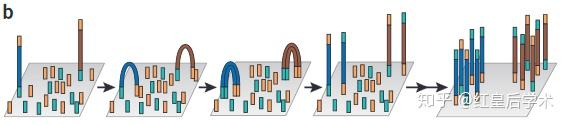
Illumina/Solexa技术原理
1. Preparation
通过不同的方法将打碎的DNA碎片末端连接序列已知的接头,构建单链DNA测序文库。
2. Immobilization and Bridge PCR
将测序文库的每一条单链DNA通过特异性的接头固定在一个固体支撑体上,固体支撑体的每一个单独小空间中只包含一条DNA链,之后通过PCR特异性的将模板DNA进行富集,从而达到测序所需的模板量。
3. Sequencing
对每一个单独的链进行碱基互补配对,反应试剂清洗和成像捕捉,不断反复进行此三步循环,每一个循环按顺序测定序列中的一个碱基。
第二代测序技术的优缺点
第二代测序技术的优点:
- 一次能够同时得到大量的序列数据,相比于一代测序技术,通量提高了成千上万倍;
- 单条序列成本非常低廉。
第二代测序技术的缺点:
- 序列读长较短,Illumina平台最长为250-300bp,454平台也只有500bp左右;
- 由于建库中利用了PCR富集序列,因此有一些含量较少的序列可能无法被大量扩增,造成一些信息的丢失,且PCR过程中有一定概率会引入错配碱基;
- 想要得到准确和长度较长的拼接结果,需要测序的覆盖率较高,导致结果错误较多和成本增加。
第二代测序技术的应用
二代测序是现阶段科研市场的主力平台,主要应用包括:基因组测序、转录组测序、群体测序、扩增子测序、宏基因组测序、重测序等。
由于成本较低,二代测序在医学领域应用也十分广泛,主要包括:癌症基因组、遗传病基因组、肿瘤与代谢疾病等。
不同测序平台的参数比较


<hr/>第三代测序技术
以PacBio公司的SMRT技术和Oxford Nanopore Technologies公司的纳米孔单分子技术为代表的新一代测序技术被称为第三代测序技术,与前两代测序技术相比,其最大的特点就是单分子测序,测序过程无需进行PCR扩增,并且理论上可以测定无限长度的核酸序列。
PacBio技术平台
SMRT芯片是一种带有很多ZMW孔的厚度为100nm的金属片,将DNA聚合酶、待测序列和不同荧光标记的dNTP放入ZMW孔的底部。荧光标记的位置是磷酸基团,当一个dNTP被添加到合成链上的同时,它会进入ZMW孔的荧光信号检测区,根据荧光的种类就可以判定dNTP的种类,从而获得核酸的碱基序列信息。
ZMW孔
PacBio平台测序原理
每个ZWM孔只允许一条DNA模板进入,DNA模板进入后,DNA聚合酶与模板结合,加入4种不同颜色荧光标记4种dNTP,其通过布朗运动随机进入检测区域并与聚合酶结合从而延伸模板,与模板匹配的碱基生成化学键的时间远远长于其他碱基停留的时间,因此统计荧光信号存在时间的长短,可区分匹配的碱基与游离碱基。通过统计4种荧光信号与时间的关系,即可测定DNA模板序列。
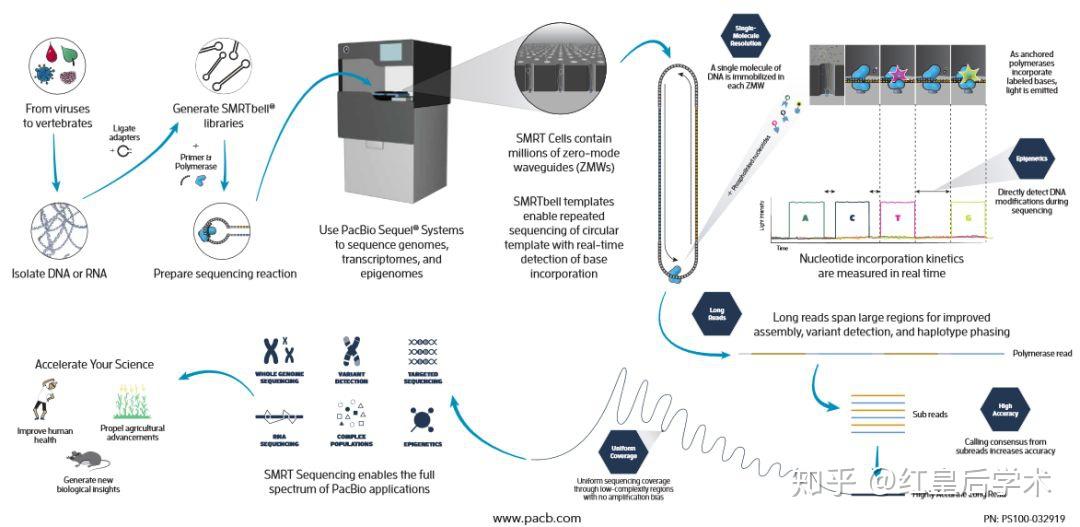
PacBio测序原理(来自PacBio官网)
PacBio平台技术关键
- DNA聚合酶,该技术得到的序列读长主要跟DNA聚合酶的活性有关,它主要受激光对其造成的损伤所影响。
- 荧光基团标记在核苷酸3'端磷酸上,在DNA合成过程中,3'端的磷酸键随着DNA链的延伸被断开,标记物被弃去,减少了DNA合成的空间位阻,维持DNA链连续合成,延长了测序读长。
- ZMW (零模波导孔),将反应信号与周围游离碱基的强大荧光背景进行区分,在一个反应管中有许多这样的圆形纳米小孔,其外径仅有100nm,激光从底部打出后不能穿透小孔进入上方溶液区,能量被限制在一个小范围里,使得荧光信号仅来自这个小反应区域,孔外其它游离核苷酸单体依然留在黑暗中,从而实现将背景荧光降到最低。
PacBio平台技术优势
1. 近乎完美的一致性和准确性
三代测序单碱基错误率虽然很高,但是这种单碱基的错误是随机发生的,因此,对同一段序列测序覆盖多次就能够进行纠错,一般覆盖到10X以上的深度就能达到99.9%的正确率。
2. 不存在测序的偏好性
因为SMRT技术在样本制备时无需PCR扩增,对于某些具有极端的碱基组成的核酸区域,三代测序也是无偏好性的,同时也不受回文序列的影响。
3. 序列准确比对
二代测序得到的序列由于长度不够,在进行比对时,会出现很多错误匹配,从而造成假阳性SNP位点;而PacBio测序平台得到的序列能够较均匀的覆盖参考基因组,每个序列能够明确的比对到相应的区域,在避免假阳性的同时,得到更加准确的变异位点和类型。
PacBio技术的优缺点
PacBio技术的优点:
- 无需PCR扩增,不会人为的引入突变;
- 超长读长,平均读长可达到10Kb,最长读长可以达到40Kb;
- 覆盖均匀,无GC偏好性;
- 通过reads的自我矫正,10X以上准确率能够达到99.9%;
- 可以直接检测到甲基化信息,同步进行表观遗传学识别。
PacBio技术的缺点:
- 单条序列错误率较高,平均核苷酸准确性不到85%;
- 测序成本较贵。
PacBio技术的应用
基因组组装
利用PacBio测序平台,可以克服部分序列GC含量高或重复序列多等问题,更好的进行基因组详细描绘,从而进行精细的基因注释等研究。
PacBio测序平台不需要进行PCR扩增,因此可以减少基因组组装过程中的人为错误和偏差。
PacBio测序平台读长较长,因此相比二代测序拼接结果更为准确,同时可以利用其长片段来填补二代数据组装中产生的gap和连接contig为scaffold。
全长转录组测序
利用PacBio测序平台读长较长的特点,进行转录组测序可以直接得到转录本的全长序列,省去了二代测序的拼接过程,使得过程更为简便,结果更为准确。
甲基化分析
PacBio测序的技术原理可以直接检测到发生甲基化的核苷酸,因此可以在进行其它测序分析的同时完成DNA甲基化的分析。
Nanopore技术测序原理
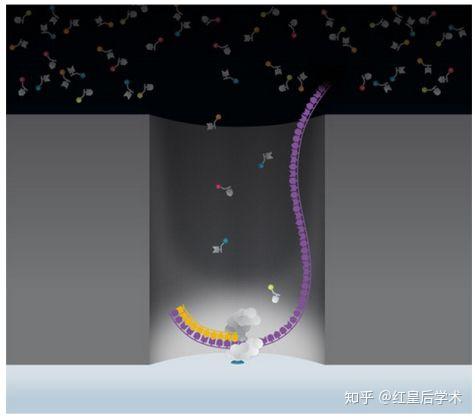
将在某一面上含有一对电极的特殊脂质双分子层置于一个微孔之上,该双分子层中含有很多由α溶血素蛋白组成的纳米孔,并且每个纳米孔会结合一个核酸外切酶。
当DNA模板进入孔道时,孔道中的核酸外切酶会“抓住”DNA分子,顺序剪切掉穿过纳米孔道的DNA碱基,每一个碱基通过纳米孔时都会产生一个阻断,根据阻断电流的变化就能检测出相应碱基的种类,最终得出DNA分子的序列。
Nanopore技术原理
Nanopore技术的优缺点
Nanopore技术的优点:
- 可以检测结构变异和可变剪切;
- 能直接对RNA分子进行测序;
- 能对修饰过的碱基进行测序;
- 测序读长更长,可以达到150kb;
- 测序数据可以做到实时监控;
- 运行速度快。
Nanopore技术的缺点:
采用的是水解测序法,不能进行重复测序,因而无法达到一个满意的测序精确度。
Nanopore技术的应用
基因组组装
利用其测序长的特点,可以填补基因组中大片段的gap。
临床应用
对于临床实践,实时获取和分析DNA/RNA序列是一件很重要的事情,对于传统的高通量测序,做到这一点非常困难,但对于Nanopore技术平台,实现实时获取序列相对容易。
Nanopore技术平台体积小、易操作,其通过电流变化检测并识别碱基,这种设计允许用户在测序过程中根据实时结果做出一些判断,对于即时诊疗有重要意义。
基于Nanopore技术的测序平台获得序列时间相比于其它平台要短得多,因此更加适合于临床环境,使得医生能够快速获得检测结果。利用Nanopore技术平台从临床样品准备到发现致病菌只需要6小时,而完成胎儿非整倍体检查只需4小时,这些应用如果利用二代测序平台可能需要数天甚至数周时间。
甲基化分析
Nanopore测序技术可以检测四种胞嘧啶碱基修饰,分别为5-methycytosine、5-hydroxymethycytosine、5-formylcytosine和5-carboxylcytosine,检测准确率为92%-98%。
不同三代测序平台的比较

<hr/>图片来源:
Shendure J, Ji, Hanlee. Next-generation DNA sequencing[J]. NATURE BIOTECHNOLOGY, 2008, 26(10):1135-1145.
Zhang Z, Shen J, Wang H, et al. Effects of Graphene Nanopore Geometry on DNA Sequencing[J]. Journal of Physical Chemistry Letters, 2014, 5(9):1602–1607.
写在后面:该篇文章并非完全原创,是多年前整理网络上相关文章而得,当时只是留作自学用途,但是由于时间较长,具体参考了那些文章已经忘记了,在此对这些文章的作者深表感谢!
关注公众号“红皇后学术”,获得更多分子生物学和组学研究知识。

原文地址:https://zhuanlan.zhihu.com/p/77663085 |
|

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 






 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 发表于 2024-9-28 08:49
发表于 2024-9-28 08:49



