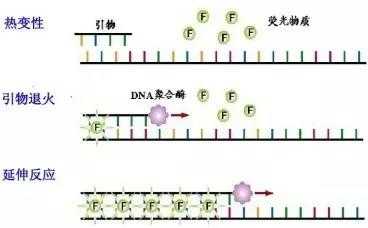
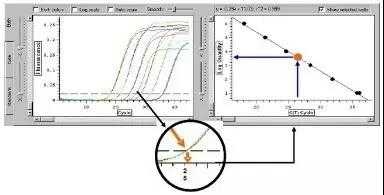
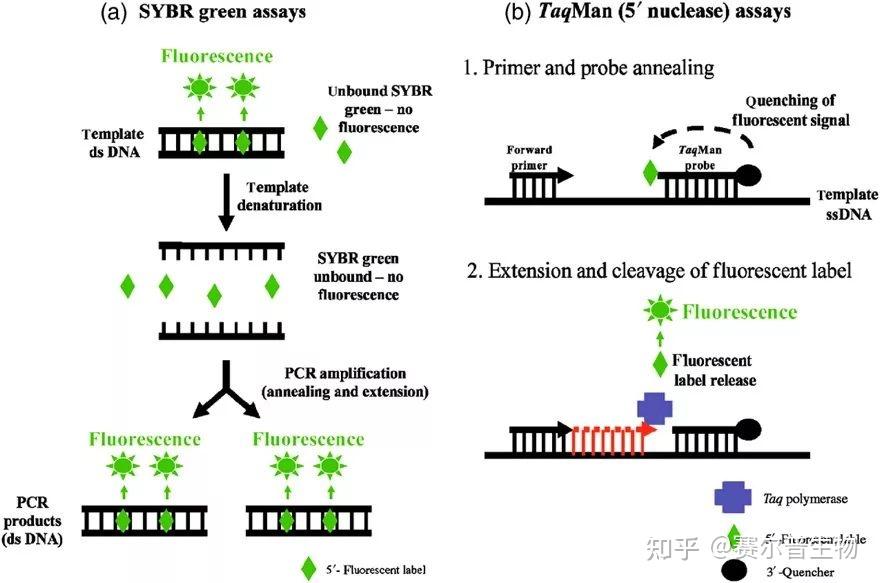
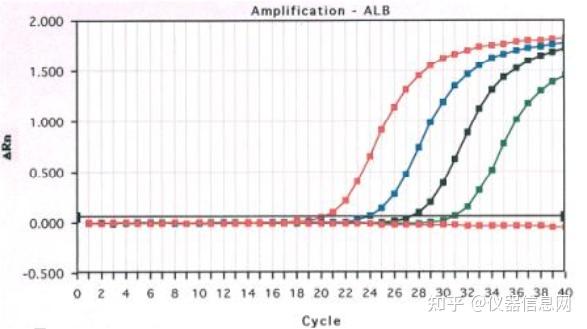
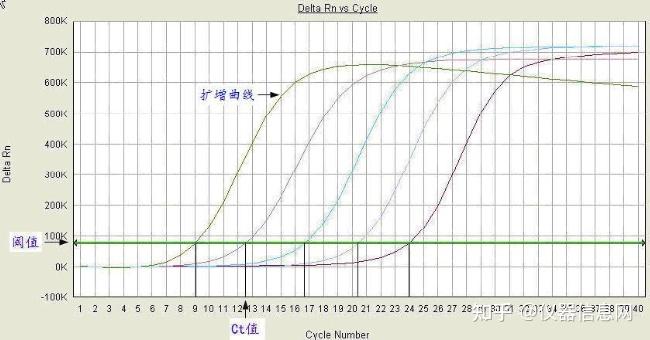
荧光染料法是在普通 PCR 反应体系中加入过量荧光染料。荧光染料在游离状态下基本不发光,与双链 DNA 结合后才释放出荧光信号。因此,在 PCR 体系中,随着特异性 PCR 产物的扩增,染料掺入双链 DNA 而产生的信号强度与 PCR 产物的数量是呈正相关的。
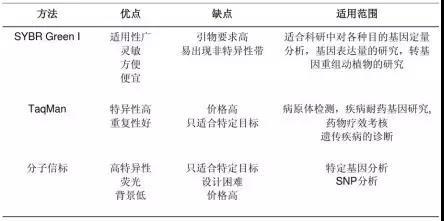
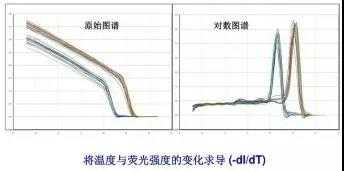
荧光染料包括饱和荧光染料和非饱和荧光染料。目前最常用的染料分子 SYBR Green I 属于非饱和荧光 染料,Eva Green 和 Solis Green 属于饱和荧光染料。两者的区别是在将 PCR 产物从 60°C逐渐升温到 95°C 制作溶解曲线的过程中,非饱和荧光染料会从已解开的单链上脱落,结合到临近的尚未解链的双链中继续 发光。饱和荧光染料不会重新与尚未解链的双链 DNA 结合,所以使用饱和染料制作的溶解曲线分辨率更高。
荧光染料法的优点是,实验设计简单,无需合成探针,降低了检测成本;操作简便,可用于监测任何 双链 DNA 序列的扩增。
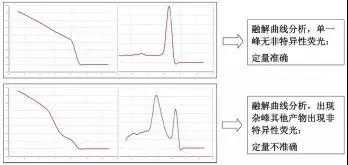
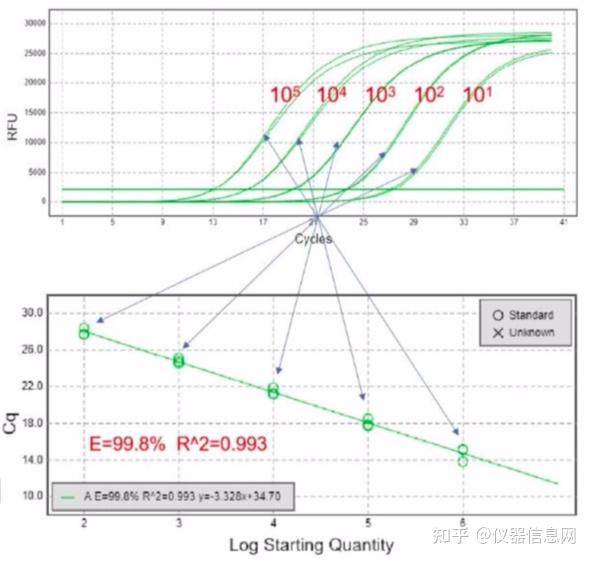
荧光染料法的最大缺点在于可能会产生假阳性信号。由于荧光染料可以与任何双链 DNA 结合,无法区分不同的双链 DNA,因此非特异产物和引物二聚体的存在都会影响检测结果的准确。通过溶解曲线,虽然可以在一定程度上对双链核酸的均一性进行检测,但是精准定量分析一般还是不用荧光染料法,而使 用荧光探针法。
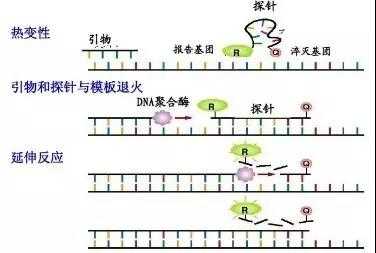
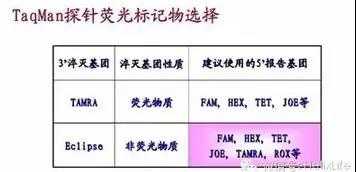
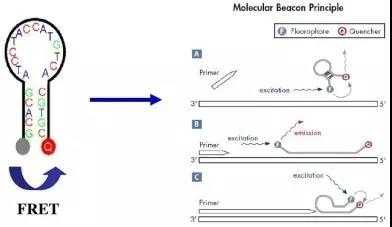
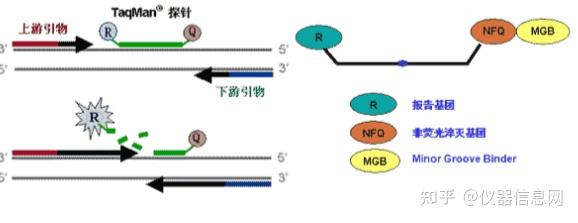
(二)荧光探针法

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 






 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 发表于 2024-9-25 20:18
发表于 2024-9-25 20:18






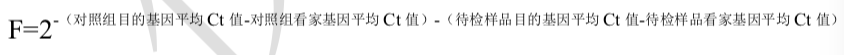


 发表于 2024-9-25 20:21
发表于 2024-9-25 20:21