只需一步,快速开始
微信扫一扫,快速登录
您需要 登录 才可以下载或查看,没有账号?立即注册
如有转载,请注明出处!!! 如有转载,请注明出处!!! 如有转载,请注明出处!!!
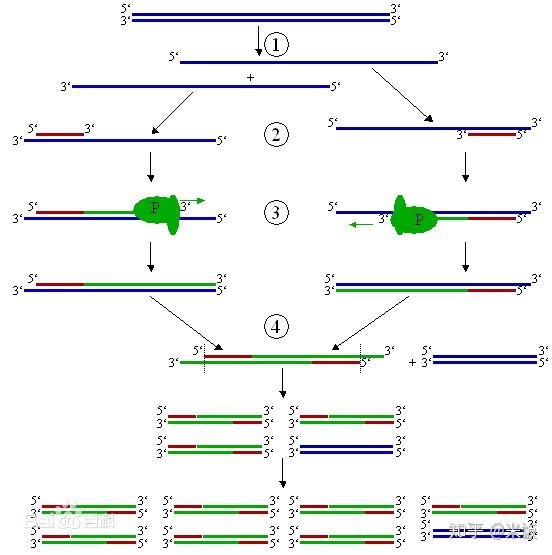
PCR技术的基本原理类似于DNA的天然复制过程,其特异性依赖于与靶序列两端互补的寡核苷酸引物。
其中dNTP、引物、模板DNA、Taq DNA聚合酶以及Mg2+的加量(或浓度)可根据实验调整,以上表格只提供大致参考值。
参考文献 1. 聚合酶链式反应和基因芯片技术的研究及在主要水生动物病毒检疫和监测中的应用 .中国知网[引用日期2019-06-20] 2. PCR之父、诺奖得主穆利斯去世,他将生物学划分为两个时代 .新浪[引用日期2019-08-10] 3. 多重聚合酶链式反应技术研究和应用 .中国知网[引用日期2019-06-20] 4. 聚合酶链式反应技术及应用 .中国知网[引用日期2019-06-20] 5. 基于连接—聚合酶链式反应定量分析RNA中6-甲基腺嘌呤 .中国知网[引用日期2019-06-20] 6. 套式聚合酶链式反应诊断三日疟原虫感染的临床应用 .中国知网[引用日期2019-06-20]
举报
本版积分规则 发表回复 回帖后跳转到最后一页
查看 »
微信扫一扫关注本站公众号

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 






 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 发表于 2024-9-24 21:55
发表于 2024-9-24 21:55