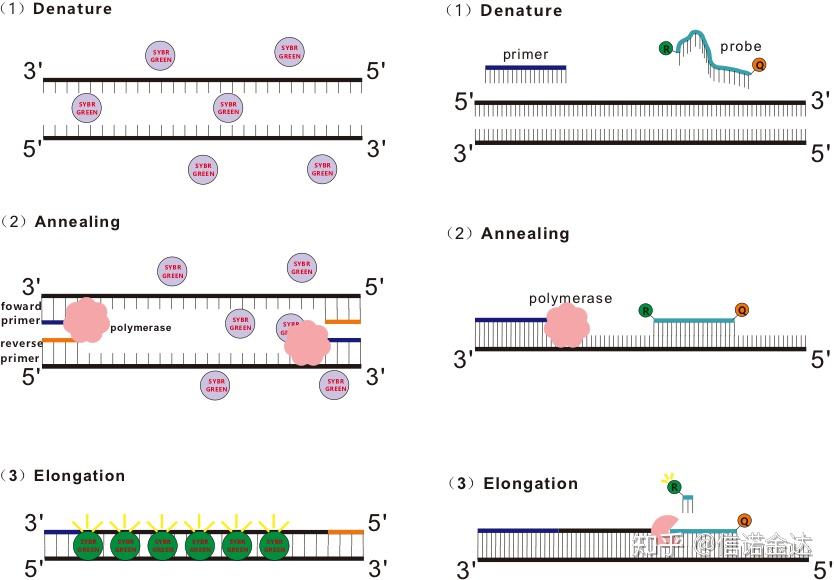
荧光染料法:
一些荧光染料如SYBR Green Ⅰ,PicoGreen,BEBO等,它们本身不发光,但结合于dsDNA的小沟后会发出荧光。所以当PCR反应刚开始时机器并不能检测到荧光信号,当反应进行到退火-延伸(二步法)或者延伸阶段(三步法),此时双链打开,在DNA聚合酶的作用下新链合成,荧光分子就结合于dsDNA的小沟中并发出荧光,随着PCR循环数的增加越来越多的染料与dsDNA结合,荧光信号也不断的增强。以SYBR Green Ⅰ为例。
探针法:
Taqman探针是最为常用的一种水解探针,在探针的5’端存在一个荧光基团,通常为FAM,探针本身则为一段与目的基因互补的序列,在探针的3’端有一个荧光猝灭基团,根据荧光共振能量转移原理(Förster resonance energy transfer, FRET),当报告荧光基团(供体荧光分子)和猝灭荧光基团(受体荧光分子)激发光谱重叠且距离很近时(7-10nm),供体分子的激发可以诱发受体分子发荧光,而自身荧光减弱。所以PCR反应开始,探针游离于体系中完整存在时,报告荧光基团并不会发出荧光,当退火时,引物和探针结合于模板,在延伸阶段,聚合酶不断的合成新链,由于DNA聚合酶具有5’-3’核酸外切酶活性,到达探针时,DNA聚合酶就会将探针从模板上水解下来,报告荧光基团和猝灭荧光基团分开,释放荧光信号。由于探针和模板存在一对一的关系,所以在试验的精度和灵敏度上,探针法都要优于染料法。
扩增效率检测:
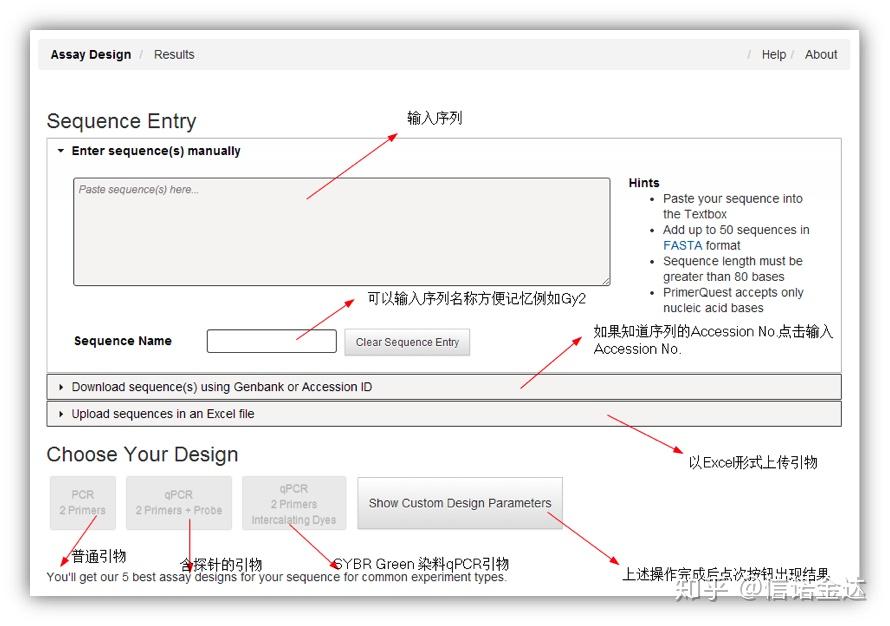
PCR反应的扩增效率严重的影响到PCR的结果,同样在qRT-PCR中,扩增效率对定量的结果尤为重要,除去反应液(reaction buffer)中其他物质及机器和protocol带来的影响,引物的质量对于qRT-PCR的扩增效率影响也非常大,为了保证结果的准确性,无论是相对荧光定量还是绝对荧光定量均需要对引物的扩增效率进行检测,而公认的有效的qRT-PCR的扩增效率为85%-115%之间有两种方法: 1.标准曲线法:
a.将cDNA混合
b.梯度稀释
c.qPCR
d.线性回归方程计算扩增效率 2.LinRegPCR
LinRegPCR is a program for the analysis of real time RT-PCR Data,also called quantitative PCR(qPCR)data based on SYBR Green or similar chemistry. The program uses non-baseline corrected data,performs a baseline correction on each sample separately,determines a window-of-linearity and then uses linear regression analysis to fit a straight line through the PCR data set. From the slope of this line the PCR efficiency of each individual sample is calculated. The mean PCR efficiency per amplicon and the Ct value per sample are used to calculate a starting concentration per sample,expressed in arbitrary fluorescence units. Data input and output are through an Excel spreadsheet.
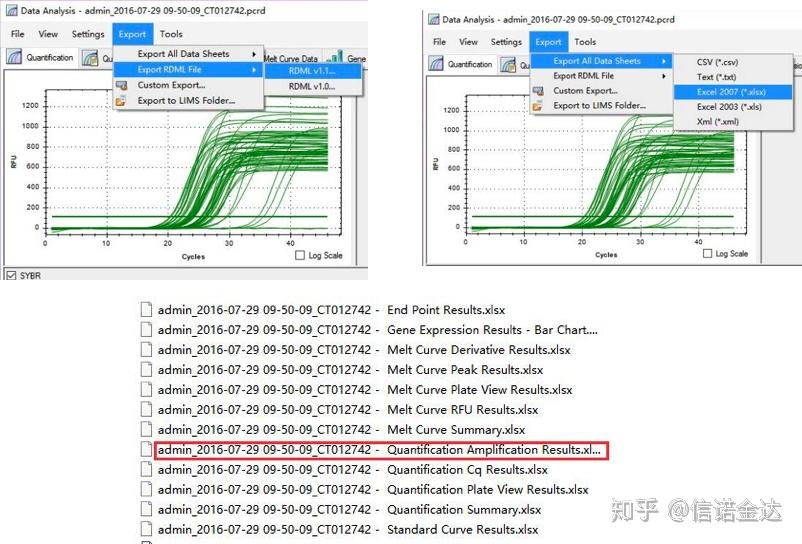
只需要混样,不需要做梯度 步骤: (以伯乐CFX96为例,不太清楚ABI的机器) 实验:为标准qPCR实验。 qPCR 数据输出 : LinRegPCR 可以识别两种形式的输出文件形式:RDML或者quantification Amplification result.实际上就是机器对循环数和荧光信号的实时检测值,通过对线性区段的荧光变化值分析得出扩增效率。 数据选择:理论上RDML值应该是可以用的,估计是我电脑的问题软件并不能识别RDML,所以我已excel输出值作为原始数据,建议先对数据进行粗筛选,例如加样失败的等造成的点可以在输出的数据中删掉(当然不删也可以,LinRegPCR后期会对这些点进行忽略)
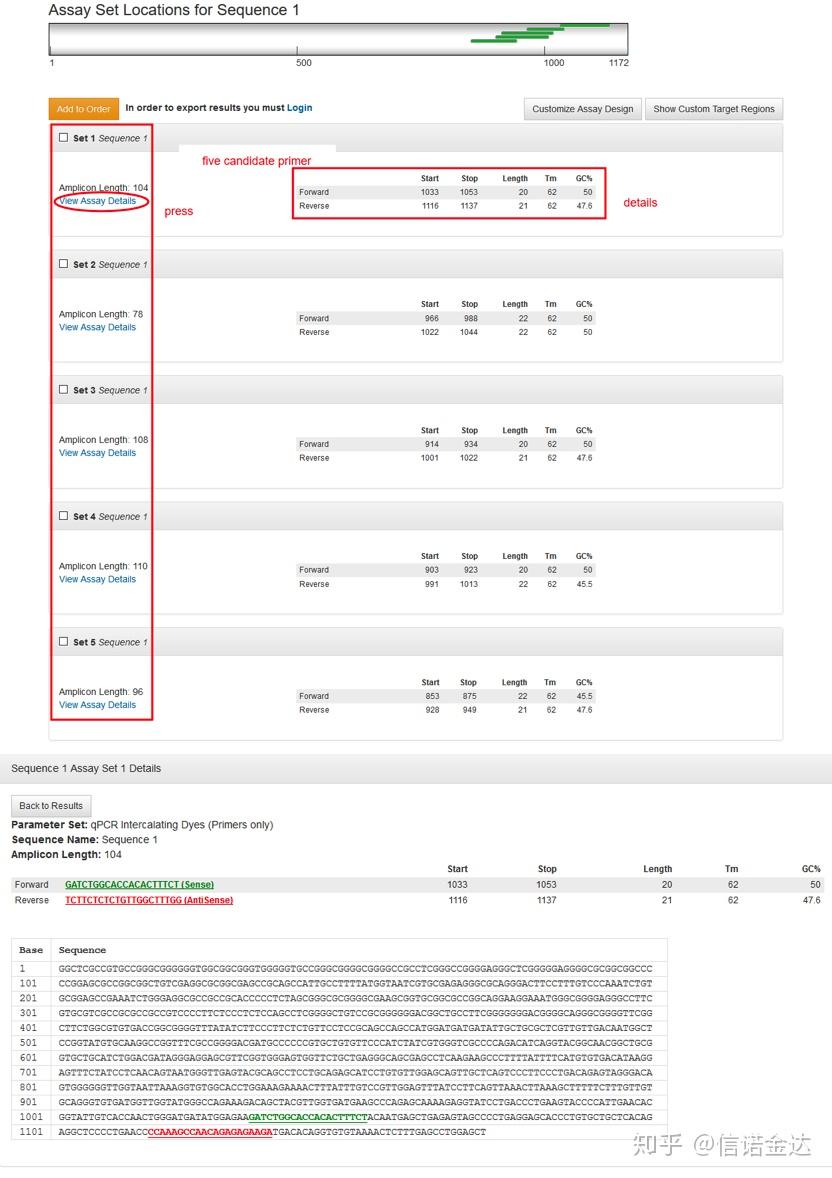
Fig5 qPCR data export
Fig6 selection of candidate samples
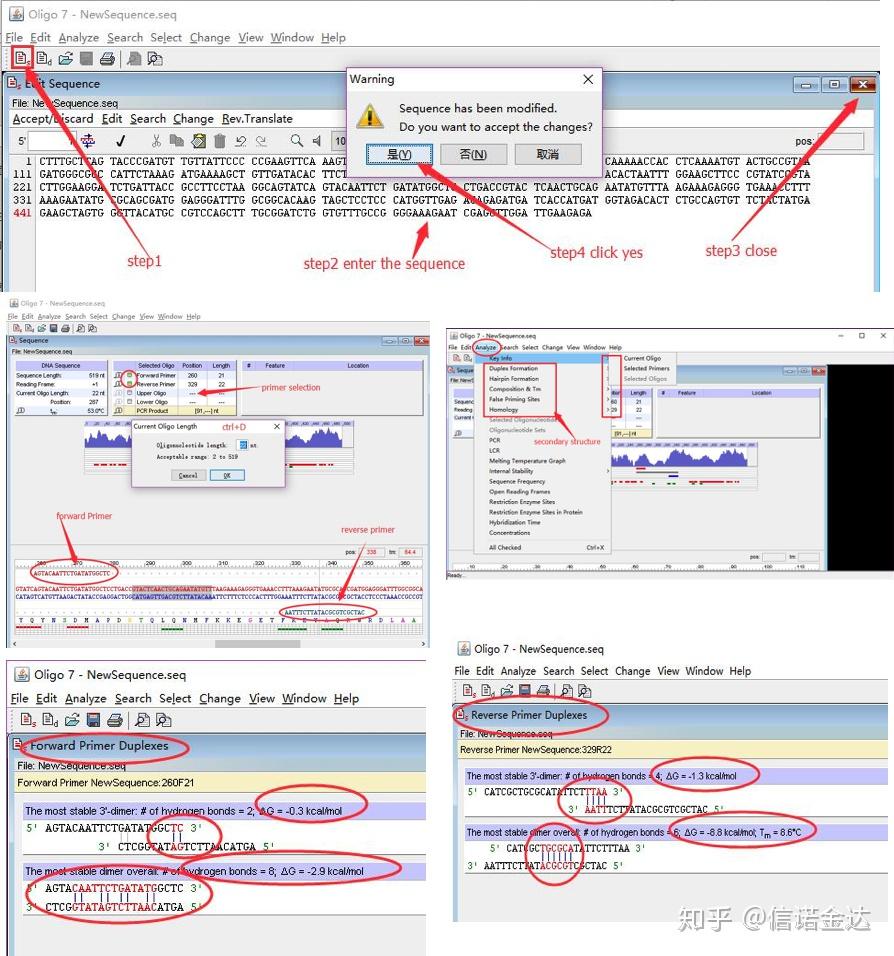
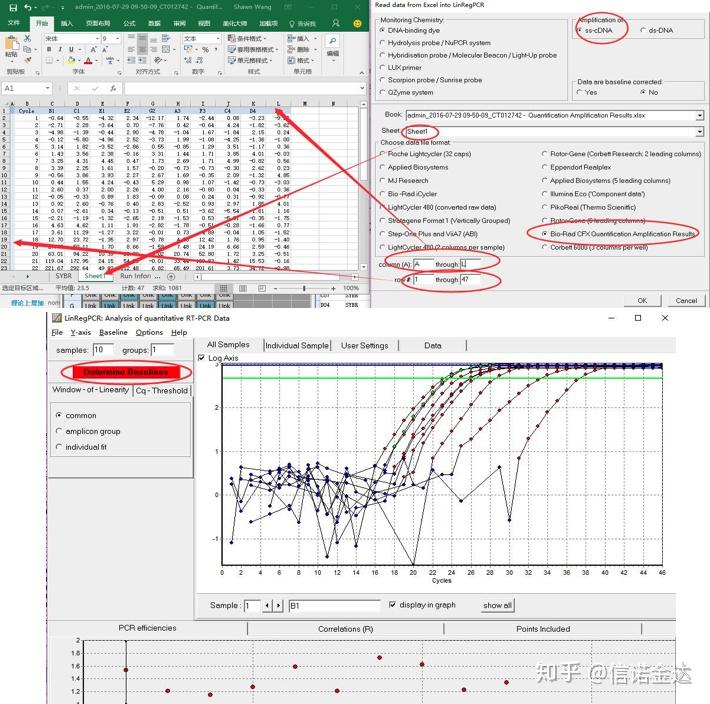
数据输入:打开 qualification amplification results. xls, →打开 LinRegPCR →文件→ read from excel → 如图7选择参数→ OK → 点击determine baselines

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 






 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 发表于 2024-9-23 18:52
发表于 2024-9-23 18:52