只需一步,快速开始
微信扫一扫,快速登录
您需要 登录 才可以下载或查看,没有账号?立即注册
举报
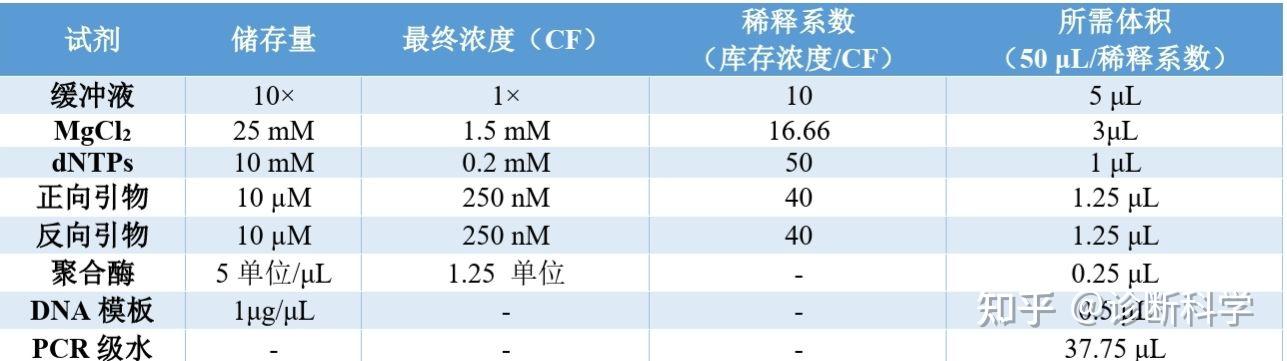
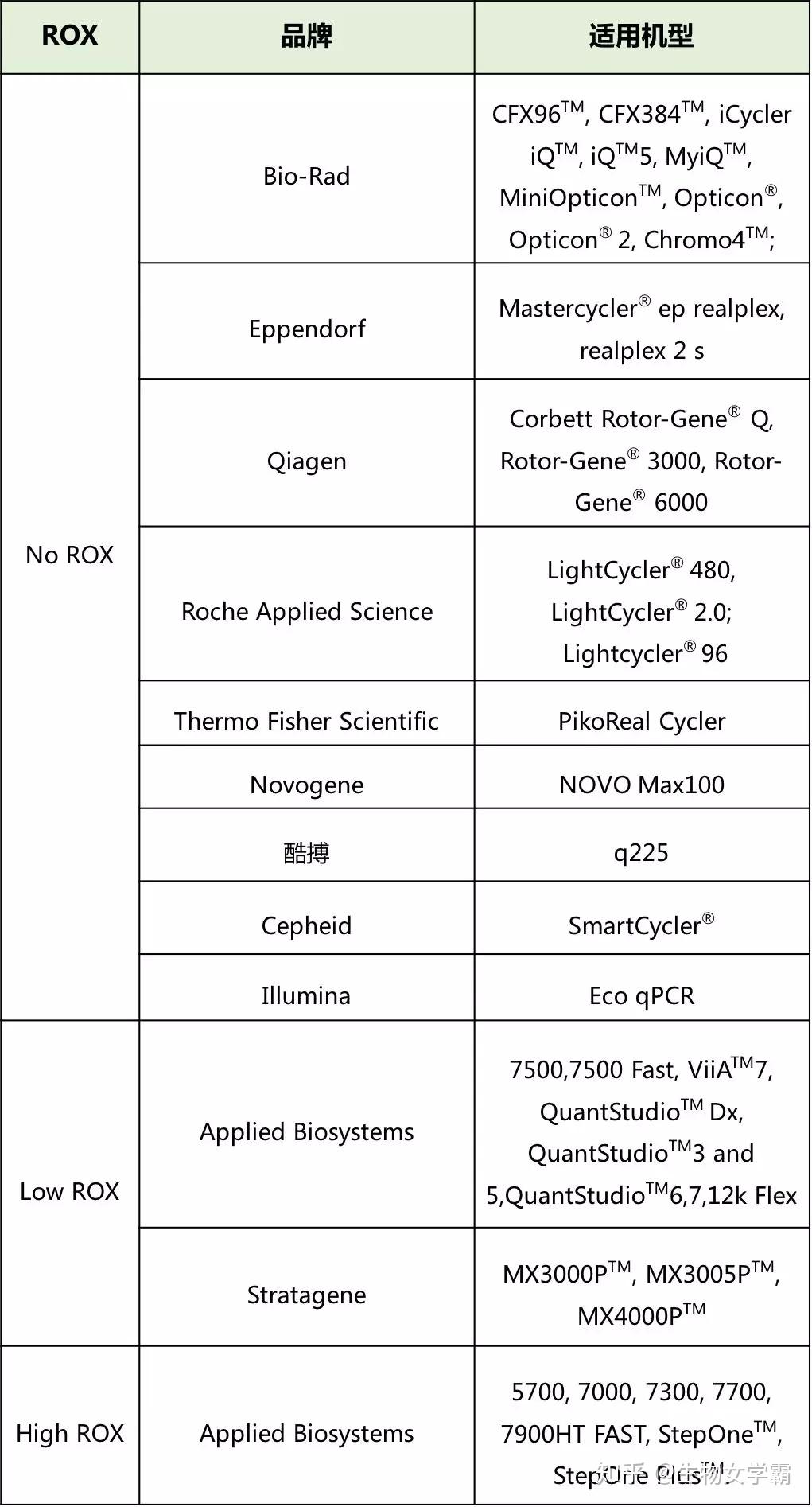
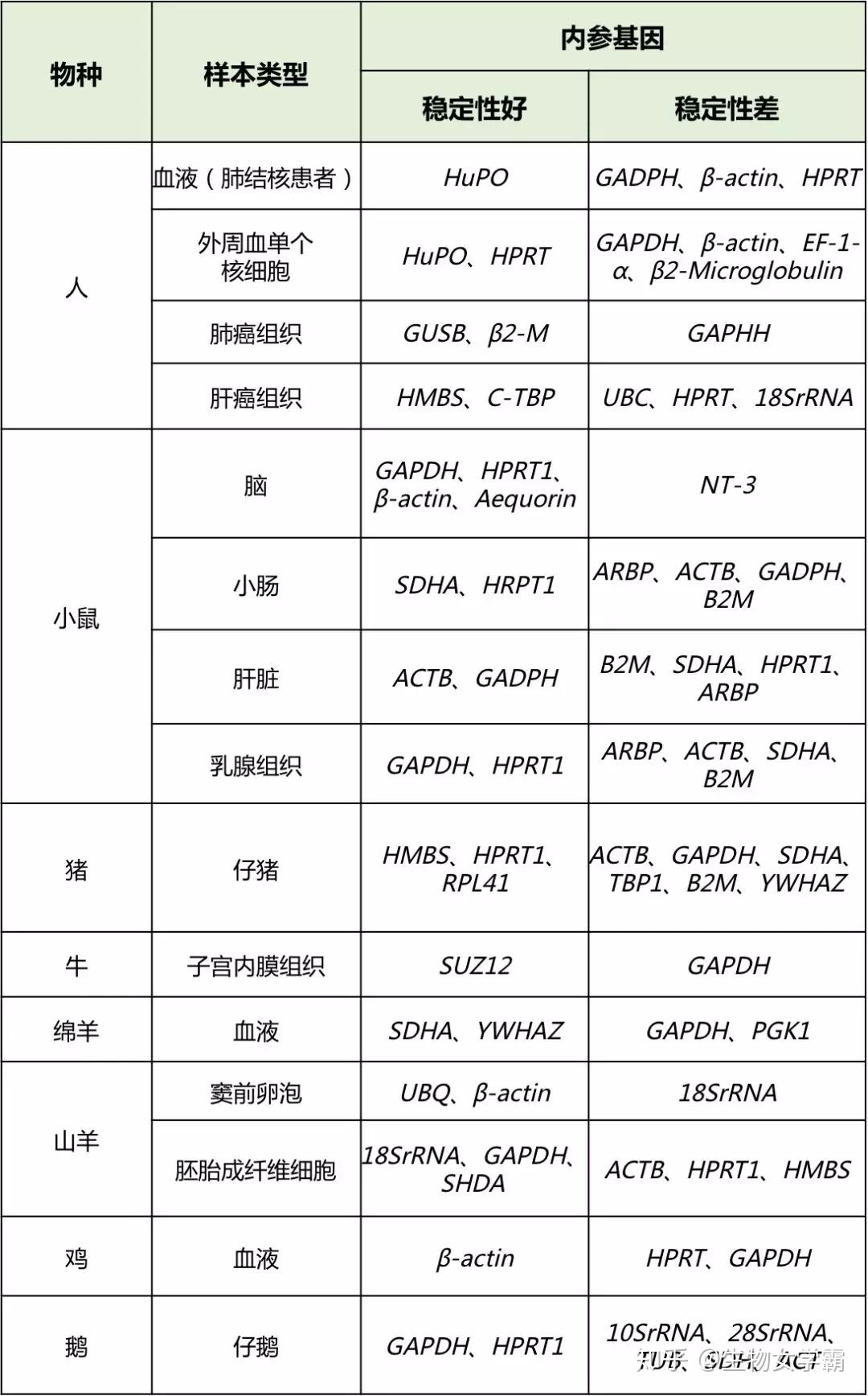
表1 | PCR Master mix混合表的例子
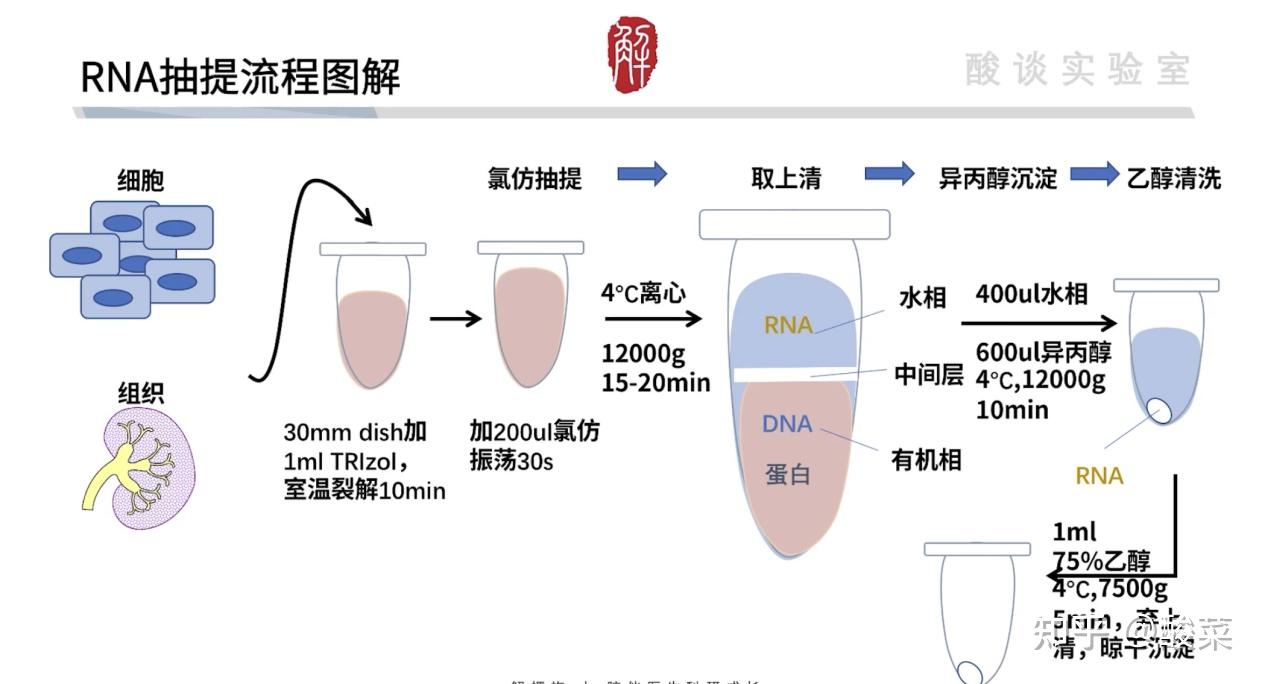
实验前期准备
实验中期操作
本版积分规则 发表回复 回帖后跳转到最后一页
查看 »
微信扫一扫关注本站公众号

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 






 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 发表于 2024-9-20 22:23
发表于 2024-9-20 22:23